Welcome to BecA bioinformatics page
To assemble a genome, computer programs typically use data consisting of single and paired reads. Single reads are simply the short sequenced fragments themselves; they can be joined up through overlapping regions into a continuous sequence known as a 'contig'. Repetitive sequences, polymorphisms, missing data and mistakes eventually limit the length of the contigs that assemblers can build.
Knowing that paired reads were generated from the same piece of DNA can help link contigs into 'scaffolds', ordered assemblies of contigs with gaps in between. Paired-read data can also indicate the size of repetitive regions and how far apart contigs are.
In this section, de novo sequence assembly guidelines are given for:
De Novo assembly using Velvet
Velvet only handles interleaved or “shuffled” fasta / fastq files, where each pair is seen one after the other, in a single file. For example:TGTTCTTATTGGACCAGAANGAGGATTTAGTGAAGAAG
+HWI-EAS210R_0001:6:1:3:663#CGATGT/1
dgggggggggfggddbb]_BTTSSSfffdddddd^`cc
@HWI-EAS210R_0001:6:1:3:663#CGATGT/2
CTACTCTCTAACAGAAAGCAGATAGTCAAACCGTGTAA
+HWI-EAS210R_0001:6:1:3:663#CGATGT/2
d`Yd`fff`dffefde^cffee^^^Ic_cceeee^WcS
shuffleSequences_fastq.pl 3kb_mp_1RC.fastq 3kb_mp_2RC.fastq 3kb_mp_shuffled.fastq
Getting the Kmer value: Default = 31. This is by default an odd number to avoid pallindromes. For the manual option you need to select a K value; this can be assisted by using the tool below, or by arbitrary testing a number of K values: http://dna.med.monash.edu.au/~torsten/velvet_advisor/
To get velvet started: To see the help message for velveth, simply run: velveth
Run the following command:
Velveth reads in these sequence files and simply produces a hashtable and two output files (Roadmaps and Sequences) which are necessary for the subsequent program, velvetg.
When we ran velveth, we specified Vtutorial as our directory name. We then specified the hash length as 31. We then specified our two input files both with -fastq -shortPaired and the file name.
When velvetg finishes it will output the number of nodes, n50, and max and total size of the assembly created. If you look in the created directory, you will also see a few files:
- contigs.fa LastGraph PreGraph Sequences
- Graph2 Log Roadmaps stats.txt
De novo assembly using CLC
CLC has a point-and-click graphical user interface and is very easy to use. The algorithm behid it is De Bruijn graphs to join reads together. More information about the the assembly algorithm works can be found hereIt is useful to try to perform several assemblies with your dataset, with varying parameter values (especially the mismatch costs), to see how the results differ.
RNA-Seq reads represent short pieces of all the mRNA present in the tissue at the time of sampling.
In order to be useful, the reads need to be combined (assembled) into larger fragments, each representing an mRNA transcript. These combined sequences are called "contigs", which is short for "contiguous sequences".
If you are working with an organism for which there is a reference genome available, you can use the gene annotations to pull out sequences coding for mRNA, and use those as the reference for further processing.
Download it in the FASTA format and skip to the section on Mapping to reference.
If not, however, you need to create your own catalog of contigs by performing a de novo assembly. A de novo assembly joins reads that overlap into contigs, while allowing a certain, user-defined, number of mismatches (variation at nucleotide positions that can be due to sequencing error or biological variation).
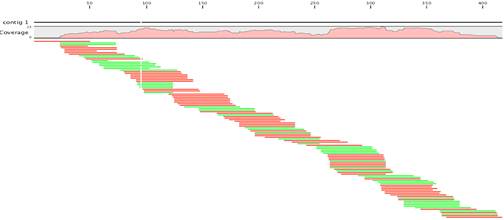
When comparing the lengths and numbers of contigs acquired from de novo assemblies to the predicted number of transcripts from genome projects, the de novo contigs typically are shorter and more numerous.
This is because the assembler cannot join contigs together unless there is enough overlap and coverage in the reads, so that several different contigs will match one mRNA transcript.
Biologically, alternative splicing of transcripts also inflates the number of contigs when compared to predictive data from genome projects. This is important to keep in mind, especially when analyzing gene expression data based on mapping to a de novo assembly. To minimize this issue, we want to use as many reads as possible in the assembly to maximize the coverage level. The assembler therefore pools the reads from all specified samples, (which means that no information about the individual samples can be extracted from the assembly). In order to get that information, we need to map our reads from each sample individually to the assembly once it has been created.
Building a de novo assembly is a very memory-intensive process. There are many programs for this, some of which are listed in the Resources page. This tutorial will guide you on how to use CLC genomics workbench, CLC is the only software in this protocol that is not open source. Luckilly, but there is one installed in the HPC server. As earlier guided, log in to the server as:
After a second or two, the CLC genomics splash screen will appear. The parameters we use in the description below is only to serve as a guide. Although they will work well for most data, it is useful to try to perform several assemblies with your dataset, with varying parameter values (especially the mismatch costs), to see how the results differ.
Objectives
The objectives of this section are to:1) import our reads into CLC,
2) build a de novo assembly,
3) examine the properties of the newly-created assembly, and
4) export our assembly from CLC. Here's also an excellent review describing and contrasting the different software packages in use:
Zhang W, Chen J, Yang Y, Tang Y, Shang J, et al. 2011.
A practical comparison of de novo genome assembly software tools for next-generation sequencing technologies. PLoS ONE 6: e17915. doi:10.1371/journal.pone.0017915
Importing data into CLC workspace
Import the quality-trimmed, adapter-clipped FASTQ files pre-processed into CLC.Note: If your reads haven't been pre-processed (adapter removal, or trimmed, follow the guidelines here)
De novo assembly
Follow these stepsMismatch cost 1. Limit 5. Uncheck "fast ungapped alignment".
Insertion and Deletion costs: 2 (no global alignment) Mismatch costs determine how many nucleotide mismatches are allowed before the reads can't be joined together. A mismatch limit of 5 allows 5 out of 50 = 10 % difference or 2 indels (as they cost 2 penalty units each).
The former prohibits ambiguities in the contigs, and instead uses the most common nucleotide. The latter option ignores all reads that match to more than one contig. As we cannot know which contig they belong to, it is safest to ignore them. Minimum contig length 200 bases.
This option makes the assembly considerably more time-intensive and can be ignored if you are pressed for time. However, the assembly can be improved by matching reads to it one extra time, and as it is very important to have as good as possible an assembly for downstream analysis, we recommend checking this option.
Export your newly created reference assembly in the FASTA format, and rename the contigs.
FASTA files contain 2 lines per sequence, one identifier line, starting with >, and one sequence line. CLC names all contigs with the name of the first input file, plus a number. We want to change the names to something simpler, such as "contig#"
Now, open your .fasta reference assembly in any text editor of choice (eg Notepad, TextWrangler, vim, etc. Note MS Word is NOT a text editor) and Find-Replace the contig names with something simpler. Make sure that the contig numbers remain, to keep each contig identifiable.
Summary
We have now created a de novo assembly, which we will use as a reference for downstream analysis.The assembly only contains information about contig sequences, and no information about how many reads were used to create them or what samples they came from. The assembly is a proxy for a library of all mRNA transcripts present in the tissue at the time of sampling, although several contigs could belong to different parts of the same mRNA molecule. In the assembly, we allowed for 5 mismatches in any one read (about 10%), ignored reads that matched to more than one contig, and set a minimum contig length of 200 bases.