Unspliced Reference mapping
Mapping refers to the process of aligning short reads to a reference sequence, whether the reference is a complete genome, transcriptome, or de novo assembly. There are numerous programs that have been developed to map reads to a reference sequence that vary in their algorithms and therefore speed (see Flicek and Birney 2009 and references therein).
In this section, we will map the reads from each of your cleaned and trimmed FASTQ files to the de novo reference assembly that you previously created (or the predicted genes if your study species has a genome sequence). Specifically, we will
1) create an index for the reference assembly (just once),
2) for each sample, map reads to the reference assembly, and
3) convert the resulting file into the SAM file format and append "read group" names to the SAM file for each sample.
Below are guideliens for reference mapping:
Overview
You will use the SAM files for your samples to extract gene expression information (the number of reads that map to each reference sequence) and to identify polymorphisms across your data.
Reference Mapping using BWA
This section will diuscuss a program that we utilize in this pipeline, called BWA (Li and Durbin 2009). It uses a Burrow's Wheeler Transform method that results in much faster processing than the first wave of programs that used a hash-based algorithm such as MAQ (Li et al. 2008).There are several parameters that can be defined for the alignment process, including:
- the number of differences allowed between reference and query (-n),
- the number of differences allowed in the seed (-k),
- the number allowed and penalty for gap openings (-o, -O), and
- the number and penalty for gap extensions (-e, -E).
To optimize the parameters for your data, i.e. to obtain the highest number of high quality mapped reads, you should generate an evaluation data file (cleaned and trimmed FASTQ file from a single sample) that you can use to run through several permutations of bwa, changing a single parameter each time. You can use the counts script from the gene expression section of this pipeline to see the number and quality of reads mapped with each set of parameters.
The number of nucleotide differences (-n) is probably the most important mapping parameter to fine-tune for your data. This should match the expected number of differences between two sequences for your species. n can be an integer (maximum edit distance, in other words the number of changes needed to convert one string to another, e.g. 5) or a fraction of missing alignments given a 2% uniform base error rate (e.g. 0.04). Counter intuitively, the larger the fraction, the fewer mismatches allowed and visa versa.
Terminology:
- Seed – a stretch of nucleotides within a read that are used to initiate the alignment process.- Phred-scale – a number scale often used for quality scores; given a p-value ranging from 0 to 1 (probability of the mapping being incorrect), the Phred score is -10*log10p, rounded to the nearest integer.
On the terminal, type bwa and press enter, to get the available options for the different analysis steps
Paired-end reads are aligned separately and then combined:
bwa aln ref readset_clean_R2.fq > readset_R2.sai
Then you need to combine the information from the two files:
samtools view -S readset_ref_bwa.sam -b -o readset_ref_bwa.bam
or to remove unmapped reads at this stage
You can open up the bam file mapped onto the reference in tablet and visualize the alignment but first you need to sort it and create and index of the bam file
samtools sort readset_ref_bwa.bam readset_ref_bwa
samtools index readset_ref_bwa.bam
Increase sensitivity of samtools using -E especially when you have multiple nucleotide polymorphisms, for example if you have virus intra-host diversity
Convert the fastq format to fasta using seqret from emboss
Reference mapping using Stampy
Stampy is another alignment tool which allows variation (particularly insertion/deletions) and thus can be used if you do not have a closely related reference. Create stampy reference, index and hash table:stampy -G ref ref.fa
stampy -g ref -H ref
Creation of BAM file and Consensus as before
samtools view -S readset_stampy.sam -b -o readset_stampy.bamsamtools sort readset_stampy.bam readset_stampy
samtools index readset_stampy.bam
bam2fastq --no-aligned --force --strict -o readset_stampy_unmapped#.fq readset_stampy.bam
seqret -osformat fasta readset_ref_stampy_E_cons.fq -out2 readset_ref_stampy_E_cons.fa
Example script
bwa aln 1000.fasta ERR244000_1.fastq > aln_sa1b.sai
bwa aln 1000.fasta ERR244000_2.fastq > aln_sa2b.sai
bwa sampe 1000.fasta aln_sa1b.sai aln_sa2b.sai ERR244000_1.fastq ERR244000_2.fastq > readset_refb_bwa.sam
samtools view -bS -o readset_refb_bwa.bam readset_refb_bwa.sam
samtools sort readset_refb_bwa.bam readset_refb_bwa.sorted
samtools index readset_refb_bwa.sorted.bam
samtools mpileup -d8000 -uf 1000.fasta readset_refb_bwa.sorted.bam | bcftools call -c | vcfutils.pl vcf2fq > cnsnew.fq
Reference mapping using Bowtie
As we have seen above, step 1 involves Build Index. The reference genome must first be "indexed" so that reads may be quickly aligned. Please be aware that if you want to use pre-indexed reference, bowtie2 indexes are different than "bowtie" indexes. To make your own from FASTA files, do the following:- Download FASTA files for the unmasked genome of interest if you haven't already (eg. from UCSC). Do NOT use masked sequences. Concatenate FASTA files into a single file. We can do this using the UNIX cat command, which merges files together
- From the directory containing the genome.fa file, run the "bowtie2-build" command. The default options usually work well for most genomes. For example, for hg19:
- Align sequences with bowtie (perform for each experiment): The most common output format for high-throughput sequencing is FASTQ format, which contains information about the sequence (A,C,G,Ts) and quality information which describes how certain the sequencer is of the base calls that were made. In the case of Illumina sequencing, the output is usually a "s_1_sequence.txt" file. More recently the Illumina pipeline will output a file that is debarcoded with your sample name such as "Experiment1.fastq". In addition, much of the data available in the SRA, the primary archive of high-throughput sequencing data, is in this format.
To use bowtie2 to map this data, run the following command:
bowtie2 -p <# cpu> -x
Reference mapping using CLC genomics workbench
CLC has a point-and-click graphical user interface and is very easy to use. The algorithm behid it is De Bruijn graphs to join reads together. More information about the the assembly algorithm works can be found hereIt is useful to try to perform several assemblies with your dataset, with varying parameter values (especially the mismatch costs), to see how the results differ.
After a second or two, the CLC genomics splash screen will appear. The parameters we use in the description below is only to serve as a guide.
Objectives
The objectives of this section are to:1) import our reads into CLC,
2) Map the reads to a reference sequence,
3) examine the properties of the newly-created assembly, and
4) export our assembly from CLC.
Importing data into CLC workspace
Import the quality-trimmed, adapter-clipped FASTQ files pre-processed into CLC.Note: If your reads haven't been pre-processed (adapter removal, or trimmed, follow the guidelines here)
Reference mapping
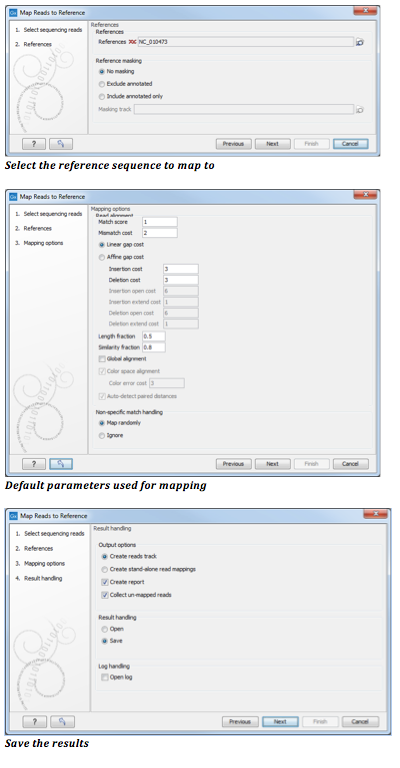
Follow these stepsClick on the "Browse" button to select the reference sequenece and click next. For starters, you can use the default mapping settings/ parameters show n on the Wizard. Click next.
Click in the radio button beside Create reads track.
Click in the box beside Create report so there is a check mark in it.
Click in the box beside Collect unmapped reads so there is a check mark in it.
Choose to Save the results. After clicking next you will select a loaction to save the mapping.
- List of non-mapped reads: These are the reads that did not match the reference sequence. You can use this list to investigate contamination in the sample or structural differences between the sequencing data and the reference sequence. Typically you will do a de novo assembly of these reads and then use BLAST to investigate the contigs
- Report: The report shows information about the mapping. Most importantly, it shows the number of reads that matched the reference sequence.
- MappingThe mapping itself shows the alignment of all the reads to the reference.
For a comprehensive guide on mapping reference, See the CLC Guide here and also here