Welcome to BecA bioinformatics page
This portal serves to introduce various bioinformatics resources and tools to aid ABCF placements analyse and intepret their data.
Simple sequence analysis
Multiple Sequence alignments
- Chimera - excellent molecular graphics package with support for a wide range of operations
- CLUSTAL
- Clustal-W - the famous Clustal-W multiple alignment program
- Clustal-X - provides a window-based user interface to the Clustal-W multiple alignment program
- DCSE - a multiple alignment editor
- Friend - an Integrated Front-end Application for Bioinformatics
- Jalview - a Java multiple alignment editor
- Mauve - a multiple genome alignment and visualization package that considers large-scale rearrangements in addition to nucleotide substitution and indels
- ModView - a program to visualize and analyze multiple biomolecule structures and/or sequence alignments.
- Musca - multiple sequence alignment of amino acid or nucleotide sequences; uses pattern discovery
- MUSCLE - more accurate than T-Coffee, faster than Clustal-W.
- SeaView - a graphical multiple sequence alignment editor
- ShadyBox - the first GUI based WYSIWYG multiple sequence alignment drawing program for major Unix platforms
- UGENE - contains multiple alignment editor with MUSCLE alignment algorithm integrated.
- BSEdit - multiple DNA/RNA/Protein Sequence Editor for Windows XP/Vista/Windows 7.
Phylogeny
- Figtree - excellent molecular graphics package with support for a wide range of operations
Advanced sequence analysis (Next generation Sequencing)
Pre processing: Sequence trimming,error correction and QC
- dupRadar dupRadar: An R package which provides functions for plotting and analyzing the duplication rates dependent on the expression levels.
- FastQC FastQC is a quality control tool for high-throughput sequence data (Babraham Institute) and is developed in Java. Import of data is possible from FastQ files, BAM or SAM format. This tool provides an overview to inform about problematic areas, summary graphs and tables to rapid assessment of data. Results are presented in HTML permanent reports. FastQC can be run as a stand-alone application or it can be integrated into a larger pipeline solution. See also seqanswers/FastQC.
- Kraken kraken: A set of tools for quality control and analysis of high-throughput sequence data.
- HTSeq HTSeq. The Python script htseq-qa takes a file with sequencing reads (either raw or aligned reads) and produces a PDF file with useful plots to assess the technical quality of a run.
- HTSeq HTSeq. The Python script htseq-qa takes a file with sequencing reads (either raw or aligned reads) and produces a PDF file with useful plots to assess the technical quality of a run.
- mRIN mRIN- Assessing mRNA integrity directly from RNA-Seq data.
- NGSQC NGSQC: cross-platform quality analysis pipeline for deep sequencing data.
- qrqc qrqc. Quickly scans reads and gathers statistics on base and quality frequencies, read length, and frequent sequences. Produces graphical output of statistics for use in quality control pipelines, and an optional HTML quality report. S4 SequenceSummary objects allow specific tests and functionality to be written around the data collected.
- RNA-SeQC RNA-SeQC is a tool with application in experiment design, process optimization and quality control before computational analysis. Essentially, provides three types of quality control: read counts (such as duplicate reads, mapped reads and mapped unique reads, rRNA reads, transcript-annotated reads, strand specificity), coverage (like mean coverage, mean coefficient of variation, 5’/3’ coverage, gaps in coverage, GC bias) and expression correlation (the tool provides RPKM-based estimation of expression levels). RNA-SeQC is implemented in Java and is not required installation, however can be run using the GenePattern web interface. The input could be one or more BAM files. HTML reports are generated as output.
- RSeQC RSeQC analyzes diverse aspects of RNA-Seq experiments: sequence quality, sequencing depth, strand specificity, GC bias, read distribution over the genome structure and coverage uniformity. The input can be SAM, BAM, FASTA, BED files or Chromosome size file (two-column, plain text file). Visualization can be performed by genome browsers like UCSC, IGB and IGV. However, R scripts can also be used to visualization.
- SAMStat SAMStat identifies problems and reports several statistics at different phases of the process. This tool evaluates unmapped, poorly and accurately mapped sequences independently to infer possible causes of poor mapping.
Improving the Quality
Improvement of the RNA-Seq quality, correcting the bias is a complex subject. Each RNA-Seq protocol introduces specific type of bias, each step of the process (such as the sequencing technology used) is susceptible to generate some sort of noise or type of error. Furthermore, even the specie under investigation and the biological context of the samples are able to influence the results and introduce some kind of bias. Many sources of bias were already reported – GC content and PCR enrichment, rRNA depletion, errors produced during sequencing, priming of reverse transcription caused by random hexamers.
Different tools were developed to attempt to solve each of the detected errors.
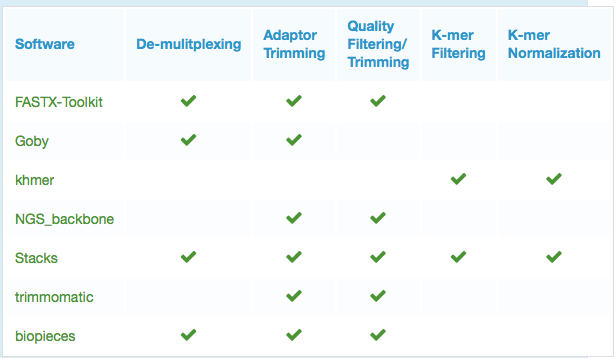
Trimming and adapters removal
- BBDuk BBDuk. Ultrafast, multithreaded tool to trim adapters and filter or mask contaminants based on kmer-matching, allowing a hamming- or edit-distance, as well as degenerate bases. Also performs optimal quality-trimming and filtering, format conversion, contaminant concentration reporting, gc-filtering, length-filtering, entropy-filtering, chastity-filtering, and generates text histograms for most operations. Interconverts between fastq, fasta, sam, scarf, interleaved and 2-file paired, gzipped, bzipped, ASCII-33 and ASCII-64. Keeps pairs together. Open-source, written in pure Java; supports all platforms with no recompilation and no other dependencies.
- clean_reads clean_reads cleans NGS (Sanger, 454, Illumina and solid) reads. It can trim bad quality regions, adaptors, vectors, and regular expressions. It also filters out the reads that do not meet a minimum quality criteria based on the sequence length and the mean quality.
- condetri condetri is a method for content dependent read trimming for Illumina data using quality scores of each base individually. It is independent from sequencing coverage and user interaction. The main focus of the implementation is on usability and to incorporate read trimming in next-generation sequencing data processing and analysis pipelines. It can process single-end and paired-end sequencing data of arbitrary length.
- cutadapt cutadapt removes adapter sequences from next-generation sequencing data (Illumina, SOLiD and 454). It is used especially when the read length of the sequencing machine is longer than the sequenced molecule, like the microRNA case.
- Deconseq Deconseq. Detect and remove contaminations from sequence data.
- Erne-Filter Erne is a short string alignment package whose goal is to provide an all-inclusive set of tools to handle short (NGS-like) reads. ERNE comprises ERNE-FILTER (read trimming and continamination filtering), ERNE-MAP (core alignment tool/algorithm), ERNE-BS5 (bisulfite treated reads aligner), and ERNE-PMAP/ERNE-PBS5 (distributed versions of the aligners).
- FastqMcf FastqMcf. Fastq-mcf attempts to: Detect & remove sequencing adapters and primers; Detect limited skewing at the ends of reads and clip; Detect poor quality at the ends of reads and clip; Detect Ns, and remove from ends; Remove reads with CASAVA 'Y' flag (purity filtering); Discard sequences that are too short after all of the above; Keep multiple mate-reads in sync while doing all of the above.
- FASTX FASTX Toolkit is a set of command line tools to manipulate reads in files FASTA or FASTQ format. These commands make possible preprocess the files before mapping with tools like Bowtie. Some of the tasks allowed are: conversion from FASTQ to FASTA format, information about statistics of quality, removing sequencing adapters, filtering and cutting sequences based on quality or conversion DNA/RNA.
- Flexbar Flexbar performs removal of adapter sequences, trimming and filtering features.
- FreClu FreClu improves overall alignment accuracy performing sequencing-error correction by trimming short reads, based on a clustering methodology.
- htSeqTools htSeqTools is a Bioconductor package able to perform quality control, processing of data and visualization. htSeqTools makes possible visualize sample correlations, to remove over-amplification artifacts, to assess enrichment efficiency, to correct strand bias and visualize hits.
- NxTrim NxTrim. Adapter trimming and virtual library creation routine for Illumina Nextera Mate Pair libraries.
- PRINSEQPRINSEQ generates statistics of your sequence data for sequence length, GC content, quality scores, n-plicates, complexity, tag sequences, poly-A/T tails, odds ratios. Filter the data, reformat and trim sequences.
- Sabre sabre. A barcode demultiplexing and trimming tool for FastQ files.
- Scythe scythe. A 3'-end adapter contaminant trimmer.
- SEECER seecer SEECER is a sequencing error correction algorithm for RNA-seq data sets. It takes the raw read sequences produced by a next generation sequencing platform like machines from Illumina or Roche. SEECER removes mismatch and indel errors from the raw reads and significantly improves downstream analysis of the data. Especially if the RNA-Seq data is used to produce a de novo transcriptome assembly, running SEECER can have tremendous impact on the quality of the assembly.
- Sickle Sickle. A windowed adaptive trimming tool for FASTQ files using quality.
- SnoWhite SnoWhite. Snowhite is a pipeline designed to flexibly and aggressively clean sequence reads (gDNA or cDNA) prior to assembly. It takes in and returns fastq or fasta formatted sequence files.
- ShortRead ShortRead is a package provided in the R (programming language) / BioConductor environments and allows input, manipulation, quality assessment and output of next-generation sequencing data. This tool makes possible manipulation of data, such as filter solutions to remove reads based on predefined criteria. ShortRead could be complemented with several Bioconductor packages to further analysis and visualization solutions (BioStrings, BSgenome, IRanges, and so on). See also seqanswers/ShortRead.
- TagCleaner TagCleaner. The TagCleaner tool can be used to automatically detect and efficiently remove tag sequences (e.g. WTA tags) from genomic and metagenomic datasets. It is easily configurable and provides a user-friendly interface.
- Trimmomatic Trimmomatic performs trimming for Illumina platforms and works with FASTQ reads (single or pair-ended). Some of the tasks executed are: cut adapters, cut bases in optional positions based on quality thresholds, cut reads to a specific length, converts quality scores to Phred-33/64.
Alignment Tools
After control assessment, the first step of RNA-Seq analysis involves alignment (RNA-Seq alignment) of the sequenced reads to a reference genome (if available) or to a transcriptome database.
Short (Unspliced) aligners
Short aligners are able to align continuous reads (not containing gaps result of splicing) to a genome of reference. Basically, there are two types: 1) based on the Burrows-Wheeler transform method such as Bowtie and BWA, and 2) based on Seed-extend methods, Needleman-Wunsch or Smith-Waterman algorithms.
The first group (Bowtie and BWA) is many times faster, however some tools of the second group, despite the time spent tend to be more sensitive, generating more reads correctly aligned.
For more, see a comparative study of short aligners
- BFAST BFAST aligns short reads to reference sequences and presents particular sensitivity towards errors, SNPs, insertions and deletions. BFAST works with the Smith-Waterman algorithm. See also seqanwers/BFAST.
- Bowtie Bowtie is a fast short aligner using an algorithm based on the Burrows-Wheeler transform and the FM-index. Bowtie tolerates a small number of mismatches. See also seqanswers/Bowtie.
- Burrows-Wheeler Aligner (BWA) BWA. BWA is a software package for mapping low-divergent sequences against a large reference genome, such as the human genome. It consists of three algorithms: BWA-backtrack, BWA-SW and BWA-MEM. The first algorithm is designed for Illumina sequence reads up to 100bp, while the rest two for longer sequences ranged from 70bp to 1Mbp. BWA-MEM and BWA-SW share similar features such as long-read support and split alignment, but BWA-MEM, which is the latest, is generally recommended for high-quality queries as it is faster and more accurate. BWA-MEM also has better performance than BWA-backtrack for 70-100bp Illumina reads. See also seqanswers/BWA.
- Short Oligonucleotide Analysis Package (SOAP) SOAP.
- GNUMAP GNUMAP performs alignment using a probabilistic Needleman-Wunsch algorithm. This tool is able to handle alignment in repetitive regions of a genome without losing information. The output of the program was developed to make possible easy visualization using available software.
- Maq Maq first aligns reads to reference sequences and after performs a consensus stage. On the first stage performs only ungapped alignment and tolerates up to 3 mismatches. See also seqanswers/Maq.
- Mosaik Mosaik. Mosaik is able to align reads containing short gaps using Smith-Waterman algorithm, ideal to overcome SNPs, insertions and deletions. See also seqanswers/Mosaik.
- NovoAlign (commercial) NovoAlign is a short aligner to the Illumina platform based on Needleman-Wunsch algorithm. Novoalign tolerates up to 8 mismatches per read, and up to 7bp of indels. It is able to deal with bisulphite data. Output in SAM format. See also seqanswers/NovoAlign.
- PerM PerM is a software package which was designed to perform highly efficient genome scale alignments for hundreds of millions of short reads produced by the ABI SOLiD and Illumina sequencing platforms. PerM is capable of providing full sensitivity for alignments within 4 mismatches for 50bp SOLID reads and 9 mismatches for 100bp Illumina reads.
- RazerS RazerS. See also seqanswers/RazerS.
- SEAL SEAL uses a MapReduce model to produce distributed computing on clusters of computers. Seal uses BWA to perform alignment and Picard MarkDuplicates to detection and duplicate read removal. See also seqanswers/SEAL.
- segemehl segemehl.
- SeqMap SeqMap. See also seqanswers/SeqMap.
- SHRiMP SHRiMP employs two techniques to align short reads. Firstly, the q-gram filtering technique based on multiple seeds identifies candidate regions. Secondly, these regions are investigated in detail using Smith-Waterman algorithm. See also seqanswers/SHRiMP.
- SMALT Smalt.
- Stampy Stampy combines the sensitivity of hash tables and the speed of BWA. Stampy is prepared to alignment of reads containing sequence variation like insertions and deletions. It is able to deal with reads up to 4500 bases and presents the output in SAM format. See also seqanswers/Stampy.
- ZOOM (commercial) ZOOM is a short aligner of the Illumina/Solexa 1G platform. ZOOM uses extended spaced seeds methodology building hash tables for the reads, and tolerates mismatches and insertions and deletions. See also seqanswers/ZOOM.
- WHAM Wham. WHAM is a high-throughput sequence alignment tool developed at University of Wisconsin-Madison. It aligns short DNA sequences (reads) to the whole human genome at a rate of over 1500 million 60bit/s reads per hour, which is one to two orders of magnitudes faster than the leading state-of-the-art techniques.
Spliced aligners
Many reads span exon-exon junctions and can not be aligned directly by Short aligners, thus specific aligners were necessary - Spliced aligners. Some Spliced aligners employ Short aligners to align firstly unspliced/continuous reads (exon-first approach), and after follow a different strategy to align the rest containing spliced regions - normally the reads are split into smaller segments and mapped independently. See also.
Aligners based on known splice junctions (annotation-guided aligners)
In this case the detection of splice junctions is based on data available in databases about known junctions. This type of tools cannot identify new splice junctions. Some of this data comes from other expression methods like expressed sequence tags (EST).
- Erange Erange is a tool to alignment and data quantification to mammalian transcriptomes. See also seqanswers/Erange.
- IsoformEx IsoformEx.
- MapAL MapAL.
- OSA OSA.
- PRADA prada. is a bwa based, known junction guided RNAseq alignment pipeline. It includes a module that identifies gene fusions.
- RNA-MATE RNA-MATE is a computational pipeline for alignment of data from Applied Biosystems SOLID system. Provides the possibility of quality control and trimming of reads. The genome alignments are performed using mapreads and the splice junctions are identified based on a library of known exon-junction sequences. This tool allows visualization of alignments and tag counting. See also seqanswers/RNA-MATE.
- RUM RUM performs alignment based on a pipeline, being able to manipulate reads with splice junctions, using Bowtie and Blat. The flowchart starts doing alignment against a genome and a transcriptome database executed by Bowtie. The next step is to perform alignment of unmapped sequences to the genome of reference using BLAT. In the final step all alignments are merged to get the final alignment. The input files can be in FASTA or FASTQ format. The output is presented in RUM and SAM format.
- RNASEQR RNASEQR. See also seqanswers/RNASEQR.
- SAMMate SAMMate. See also seqanswers/SAMMate.
- SpliceSeq SpliceSeq.
- X-Mate X-Mate.
De novo Splice Aligners
De novo Splice aligners allow the detection of new Splice junctions without need to previous annotated information (some of these tools present annotation as a suplementar option). See also De novo Splice Aligners.
- ABMapper ABMapper. See also seqanswers/ABMapper.
- BBMap BBMap Uses short kmers to align reads directly to the genome (spanning introns to find novel isoforms) or transcriptome. Highly tolerant of substitution errors and indels, and very fast. Supports output of all SAM tags needed by Cufflinks. No limit to genome size or number of splices per read. Supports Illumina, 454, Sanger, Ion Torrent, PacBio, and Oxford Nanopore reads, paired or single-ended. Does not use any splice-site-finding heuristics optimized for a single taxonomic branch, but rather finds optimally-scoring multi-affine-transform global alignments, and thus is ideal for studying new organisms with no annotation and unknown splice motifs. Open-source, written in pure Java; supports all platforms with no recompilation and no other dependencies. See also SeqAnswers/BBMap.
- ContextMap ContextMap was developed to overcome some limitations of other mapping approaches, such as resolution of ambiguities. The central idea of this tool is to consider reads in gene expression context, improving this way alignment accuracy. ContextMap can be used as a stand-alone program and supported by mappers producing a SAM file in the output (e.g.: TopHat or MapSplice). In stand-alone mode aligns reads to a genome, to a transcriptome database or both.
- CRAC CRAC propose a novel way of analyzing reads that integrates genomic locations and local coverage, and detect candidate mutations, indels, splice or fusion junctions in each single read. Importantly, CRAC improves its predictive performance when supplied with e.g. 200 nt reads and should fit future needs of read analyses.
- GSNAP GSNAP. See also seqanswers/GSNAP.
- HISAT HISAT. HISAT is a fast and sensitive spliced alignment program for mapping RNA-seq reads. In addition to one global FM index that represents a whole genome, HISAT uses a large set of small FM indexes that collectively cover the whole genome (each index represents a genomic region of ~64,000 bp and ~48,000 indexes are needed to cover the human genome). These small indexes (called local indexes) combined with several alignment strategies enable effective alignment of RNA-seq reads, in particular, reads spanning multiple exons. The memory footprint of HISAT is relatively low (~4.3GB for the human genome). We have developed HISAT based on the Bowtie2 implementation to handle most of the operations on the FM index.
- HMMSplicer HMMSplicer can identify canonical and non-canonical splice junctions in short-reads. Firstly, unspliced reads are removed with Bowtie. After that, the remaining reads are one at a time divided in half, then each part is seeded against a genome and the exon borders are determined based on the Hidden Markov Model. A quality score is assigned to each junction, useful to detect false positive rates. See also seqanswers/HMMSplicer.
- MapSplice MapSplice. See also seqanswers/MapSplice.
- PALMapper PALMapper. See also seqanswers/PALMapper.
- Pass Pass aligns gapped, ungapped reads and also bisulfite sequencing data. It includes the possibility to filter data before alignment (remotion of adapters). Pass uses Needleman-Wunsch and Smith-Waterman algorithms, and performs alignment in 3 stages: scanning positions of seed sequences in the genome, testing the contiguous regions and finally refining the alignment. See also seqanswers/Pass.
- PASSion PASSion.
- PASTA PASTA.
- QPALMA QPALMA predicts splice junctions supported on machine learning algorithms. In this case the training set is a set of spliced reads with quality information and already known alignments. See also seqanswers/QPALMA.
- RAZER RAZER:reads aligner for SNPs and editing sites of RNA.
- SeqSaw SeqSaw.
- SoapSplice SoapSplice.
- SpliceMap SpliceMap. See also seqanswers/SpliceMap.
- SplitSeek SplitSeek. See also seqanswers/SplitSeek.
- SuperSplat SuperSplat was developed to find all type of splice junctions. The algorithm splits each read in all possible two-chunk combinations in an iterative way, and alignment is tried to each chunck. Output in "Supersplat" format. See also seqanswers/SuperSplat.
- Subread Subread is a superfast, accurate and scalable read aligner. It uses the seed-and-vote mapping paradigm to determine the mapping location of the read by using its largest mappable region. It automatically decides whether the read should be globally mapped or locally mapped. For RNA-seq data, Subread should be used for the purpose of expression analysis. Subread is very powerful in mapping gDNA-seq reads as well. See also seqanswers/Subread.
- Subjunc Subjunc is a specialized version of Subread. It uses all mappable regions in an RNA-seq read to discover exons and exon-exon junctions. It uses the donor/receptor signals to find the exact splicing locations. Subjunc yields full alignments for every RNA-seq read including exon-spanning reads, in addition to the discovered exon-exon junctions. Subjunc should be used for the purpose of junction detection and genomic variation detection in RNA-seq data. See also seqanswers/Subjunc.
- TrueSight TrueSight: Self-training Algorithm for Splice Junction Detection using RNA-seq.
De novo Splice Aligners that also use annotation optionally[edit]
- MapNext MapNext. See also seqanswers/MapNext.
- OLego OLego. See also seqanswers/OLego.
- STAR STAR is an ultrafast tool that employs "sequential maximum mappable seed search in uncompressed suffix arrays followed by seed clustering and stitching procedure", detects canonical, non-canonical splices junctions and chimeric-fusion sequences. It is already adapted to align long reads (third-generation sequencing technologies) and can reach speeds of 45 million paired reads per hour per processor. See also seqanswers/STAR.
- TopHat TopHat is prepared to find de novo junctions. TopHat aligns reads in two steps. Firstly, unspliced reads are aligned with Bowtie. After, the aligned reads are assembled with Maq resulting islands of sequences. Secondly, the splice junctions are determined based on the initially unmapped reads and the possible canonical donor and acceptor sites within the island sequences. See also seqanswers/TopHat.
Other Spliced Aligners
- G.Mo.R-Se G.Mo.R-Se is a method that uses RNA-Seq reads to build de novo gene models.
- Cufflinks: assembles aligned RNA-Seq reads into transcripts, estimates their abundance and tests for differential expression and regulation (transcriptome-wide)
- CuffDiff is a program within cufflinks that compares transcript abundance between samples
- IGV: Integrated Genome Viewer- a high performance visualisation tool for interactive exploration of large integrated genomic datasets
- Tuxedo protocol (bowtie/ Tophat / cufflinks)
- Galaxy protocol
- Chipster protocol
Workflows and protocols