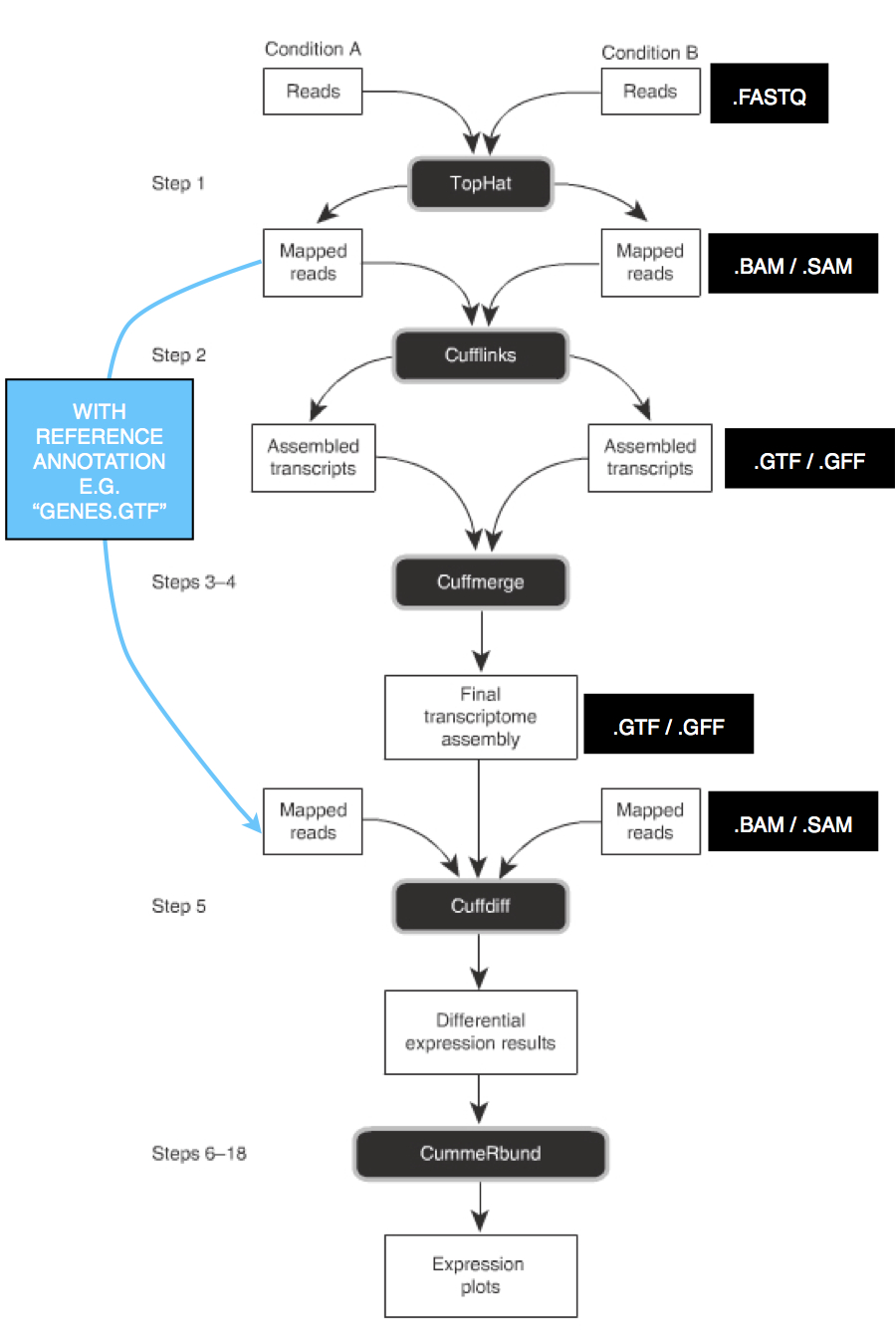
Transcriptome mapping
Mapping refers to the process of aligning short reads to a reference sequence, whether the reference is a complete genome, transcriptome, or de novo assembly. There are numerous programs that have been developed to map reads to a reference sequence that vary in their algorithms and therefore speed (see Flicek and Birney 2009 and references therein).
In this section, we will map the reads from each of your cleaned and trimmed FASTQ files to the de novo reference assembly that you previously created (or the predicted genes if your study species has a genome sequence). Specifically, we will
1) create an index for the reference assembly (just once),
2) for each sample, map reads to the reference assembly, and
3) convert the resulting file into the SAM file format and append "read group" names to the SAM file for each sample.
Below are guideliens for reference mapping:
Overview
You will use the SAM files for your samples to extract gene expression information (the number of reads that map to each reference sequence) and to identify polymorphisms across your data.
Mapping to a transcriptome reference
If are working with an organism for which there is a reference transcriptome available, you can use the gene annotations to pull out sequences coding for mRNA and use those as the reference for further processing. Download these in the FASTA format and skip to Mapping to reference. This read mapping is similar to genomic reference mapping, since the introns are non-existent in the reference transcriptome.
See reference mapping guide here
Spliced aligning (to Genomic reference) for RNA-Seq
RNA-Seq reads represent short pieces of all the mRNA present in the tissue at the time of sampling. In order to be useful, the reads need to be combined (assembled) into larger fragments, each representing an mRNA transcript. These combined sequences are called "contigs", which is short for "contiguous sequences".
Spliced assemblers are used to align sequence reads to a reference genome.
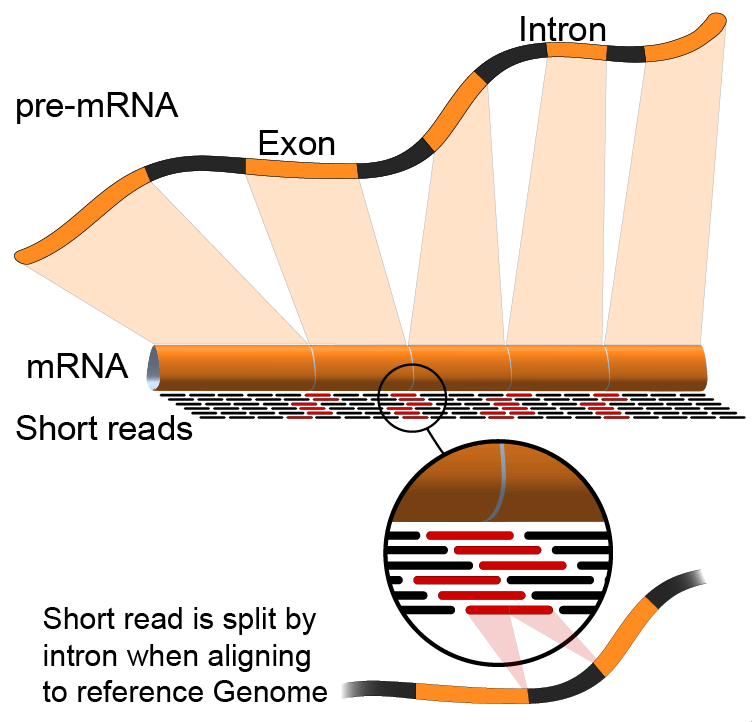
Note: Since a genome contains the sum of all introns and exons that may be present in a transcript, spliced variants that do not align continuously along the genome may be discounted as actual protein isoforms. Usually, any kind of RNA-seq method will benefit from looking for splicing junctions in addition to genomic mapping:
Transcriptome mapping using STAR
STAR is one of a growing number of short read aligners that takes advantage of advances in computational power to optimize the short read mapping process (original publication: Dobin et al.) The key limitation with STAR is computer RAM - STAR requires at least 30 Gb to align to the human or mouse genomes.- Build a genome index Like all aligners, you need to build the genome index first. First, make a directory for the index (e.g. mm9-starIndex/). Then copy the genome FASTA file into the directory and cd into it to make that directory your current directory. Then, to build the index, the command is:
- Align RNA-Seq Reads to the genome with STAR To align RNA-Seq reads with STAR, run the following command:
STAR --runMode genomeGenerate --runThreadN [# cpus] --genomeDir [genome output directory] --genomeFastaFiles [input Genome FASTA file]
i.e. STAR --runMode genomeGenerate --runThreadN 24 --genomeDir ./ --genomeFastaFiles mm9.fa
Note: For small genomes, you may need to add the following to create a proper index:
STAR --genomeDir [Directory with the Genome Index] --runThreadN [# cpus] --readFilesIn [FASTQ file] --outFileNamePrefix [OutputPrefix]
i.e. STAR --genomeDir mm9-starIndex/ --runThreadN 24 --readFilesIn Experiment1.fastq --outFileNamePrefix Experiment1Star
# paried-end data:
STAR will create several output files - the most important of which is the "*.Aligned.out.sam" - you will likely want to convert this to a bam file and sort it to use it with other programs. The default output is a SAM file.
Notes on STAR
STAR is very very fast - it will rip through 20 million reads in a matter of minutes if you have 20 cpus working for you. In fact, the longest part of the program is loading the index into memory. If you are aligning several experiments in a row, add the option "--genomeLoad LoadAndKeep" and STAR will load the genome index into shared memory so that it can use it for the next time you run the program. Also, converting the sam file into a sorted bam file will take much longer than aligning the data in the first place.
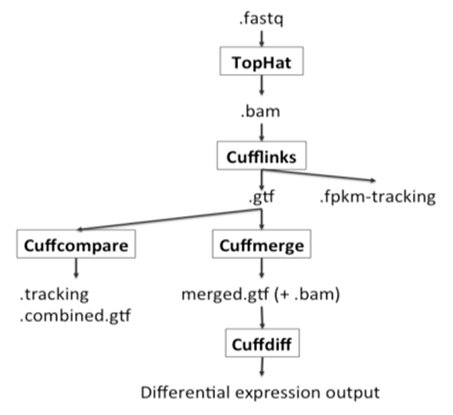
Transcriptome mapping using Tophat
Tophat is basically a specialized wrapper for bowtie2 - it manipulates your reads and aligns them with bowtie2 in order to identify novel splice junctions. It can also use given splice junctions/gene models to map your data across known splice junctions.- Build Index (takes a while, but only do this once): This part is exactly the same as for bowtie2 - if you already made or downloaded an index for bowtie2, you can skip this step.
- - Align your RNA-seq data to the genome using Tophat To use tophat to map this data, run the following command:
tophat -o TophatOutput/ -p 8 /programs/indexes/hg19 Experiment1.fastq
Paired-end Example:
tophat -o TophatOutputPE/ -p 8 /programs/indexes/hg19 Experiment1.r1.fastq Experiment1.r2.fastq
Tophat places several output files in an output directory. The most important is the "accepted_hits.bam" file - this is the alignment file that will be used for future analysis (more info here). There are additional files that can be useful, including a "junctions.bed" file with records all of the splice junctions discovered in the data.
Important Tophat Parameters:
-
--library-type [fr-unstranded | fr-firststrand | fr-secondstrand]
Describes which method was used to generate your RNA-seq library. If you used a method that doesn't produce strand specific signal (i.e. just sequencing a cDNA library), then select the fr-unstranded. If you use a stranded method that sequences the first DNA strand made (like a dUTP method), then use fr-firststrnad. If you use direct ligation methods, then fr-secondstrand. Correctly specifying the library type will help Tophat identify splice junctions.
-
-G [GTF file]
Use this option to specify a known transcriptome to map the reads against. By default, tophat will also search for de novo splice events, but this will help it known were the known ones are so that you don't miss them. A GTF files are called Gene Transfer Files, and a description of the format can be found here. To get a GTF file for your organism, you can usually get one from UCSC Table Browser.
In the output format, be sure to select GTF file - the file you download from here should work with tophat.
Tophat Mapping Strategy
If your goal is to identify novel transcripts and you have several separate experiments, it is recommended pooling all of your data together into a single expeirment/FASTQ file and mapping your data in one run. This is because one of the ways Tophat tries to identify novel junctions is by first identifying "exons" by mapping segments of reads to the genome using bowtie2. The more segments it's able to map, the more confident it is about putative exons and the greater the chance it will identify unannotated splice sites.You can then go back with your novel splice sites and remap the original experiments (not pooled) to get reads for each individual experiment using the "-j [raw junction file]". This is most useful for short reads. This general strategy is also useful if running cufflinks...