Sequence alignments
| Sequence alignments is a widely used bioinformatic technique to try to align several related sequences to find residues / bases which are conserved between sequences and which are variable. Conserved residues may be an essential part of the active site of an enzyme for example, and variable residues could be part of a 'generic' alpha helix. | 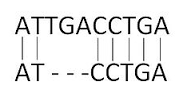 |
Click here for sequence alignment using CLC workbench
Click here for sequence alignment using Clustal W/X
The alignment of biological sequences is probably one of the most important and most accomplished in the field of bioinformatics. The ultimate goal of sequence alignment is to determine the similarity between different sequences.
Applications
- Gene finding: Having identified all ORFs of a genome can be used their lengths to estimate the probability of actually constituting genes, these are ab initio methods mentioned before. On the other hand, if there is a gene from another organism with a good alignment with the ORF found, there is strong evidence that this ORF is a gene.
- Function prediction: The alignment of sequences to determine if two genes are similar. That way if the first function of these genes is known we can assign the same function as the second. Genome Sequence Assembly: the de-novo assembly is a very important task in bioinformatics is based on the alignments between short DNA sequences obtained by the new-generation sequencers.
- One of the main applications of sequence alignment is the identification of homologous genes. Living organisms share a large number of genes descended from common ancestors and have been maintained in different organisms due to its functionality but accumulate differences that have diverged from each other. These differences may be due to mutations that change a symbol (nucleotide or amino acid) for another or insertions / deletions, indels, which insert or delete a symbol in the corresponding sequence.
There are two different forms of homology. When the origin of two homologous genes is due to a process of gene duplication within the same species these genes are called paralogs, whereas when the origin is due to a speciation process/ event resulting in homologous genes in these different species are called orthologs.
Pairwise sequence alignment
Pairwise alignments are comparison of two sequences. This can be done to determine evolutionary relationships, predict protein function and structure.To view pairwise sequence alignment, the sequences can be viewed using a dotplot matrix, where two sequences are represented along the x- and y- axis, a dot placed at the grid where the two sequences have identical residues; as shown below:
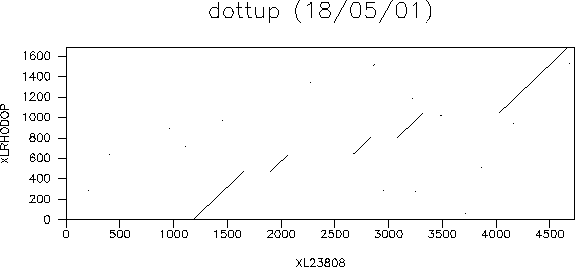
Diagonals correspond to conserved regions. A score is calculated by awarding a "reward" (positive score) for matching residues, and a penalty (negative score) for mismatches (gaps and substitutions). For example:
|
KAWRTY | ||:| KGWRSY | If Match = 5; Mismatch = 0; the score for the example shown is 5+0+5+5+0+5 = 20 |
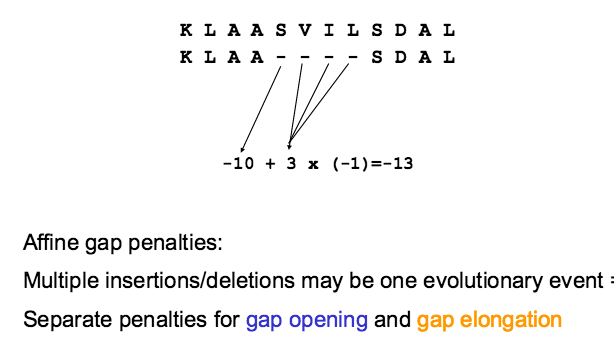
Conservative substitutions are when a residue is replaced by a residue with similar physiochemical properties. Therefore, substitutions that are conserved are less penalised from mismatches. eg Glutamate-Aspartate (Acidic amino acids) Serine-Threonine (-OH sidechain)
As a result, a matrix is prepared to give different scores of residues based on the likelihood of occurence in nature/ evolution.
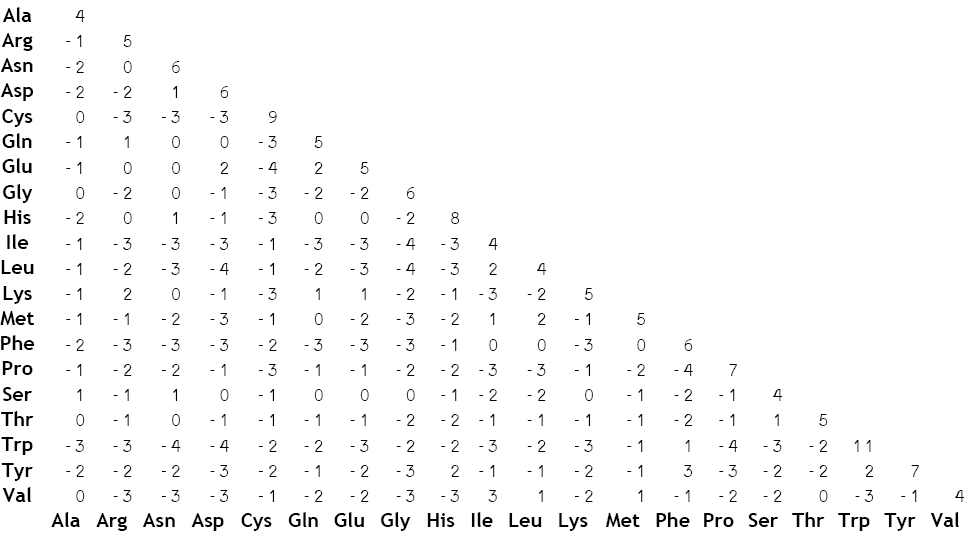
Multiple sequence alignment
Multiple sequence alignments are as a result of aligning multiple sequences. They are used to:- Identify conserved sequence regions- Conservation is indicative of functional importance
- Constructing of profiles- eg for further database searching
- Prefiction of structural features, eg alpha-helices, loops, etc.

Alignment software

Sequence alignment using CLC Main workbench
The functionalities of CLC Main Workbench are used for DNA, RNA, and protein sequence data analysis, such as gene expression analysis, primer design, molecular cloning, phylogenetic analyses, and sequence data management, amongst a wide variety of other features

Sequence alignment using CLUSTAL W/X
Go to the Clustal web page ; and click on the link that says "download here". You'll find a list of files for download. Some are for ClustalX, the graphical version of the program, and others are for ClustalW, the command line version of the program
Download the program of choice, and refer to the help pages on how to use the program you chose. Ideally, you can also refer to the program help pages by clicking on help (CLUSTALX); or typing