Introduction to Linux for Biology
| This module will enable you to navigate the UNIX environment, allowing you to create, manipulate, and store large data files efficiently. You will find that in many ways, the UNIX environment is designed for speed and simplicity in using large files in a way that is unavailable in more "user-friendly" environments like Microsoft Windows |  |
Why Linux?
- Majority of bioinformatics/computational biology software developed only for Linux
- Most programs are command line (i.e., launched by entering a command in a terminal window rather than through GUI) While various graphical and/or web user interfaces exist (e.g., Galaxy, iPlant Discovery Environment, BioHPC Web), but often struggle to provide level of flexibility needed in cutting-edge research
- Versatile scripting and system tools readily available on Linux allow customization of any analysis
Logging in to Linux server
- Request access to the server, from Alan / James (Lab 2), or ask for assistance from any of the Bioinformaticians (Mark, Joyce or Dedan)
- If you use a windows computer, you need to install an SSH client to enable you to log in to the server. Mobaxterm (preffered) or PUTTY are good options.
- Type the following on the client (Windows) / Terminal (Linux / Mac O using the login provided in (1) above
Password:
Last login: Tue Mar 18 18:10:47 2014 from 128.32.218.204
Note
The material below is intended for the in-class instruction. Others trying to learn this material on their own can follow along on any Unix (like) machine, however, please be aware the file names and path names will be different on your machine.Directory structure
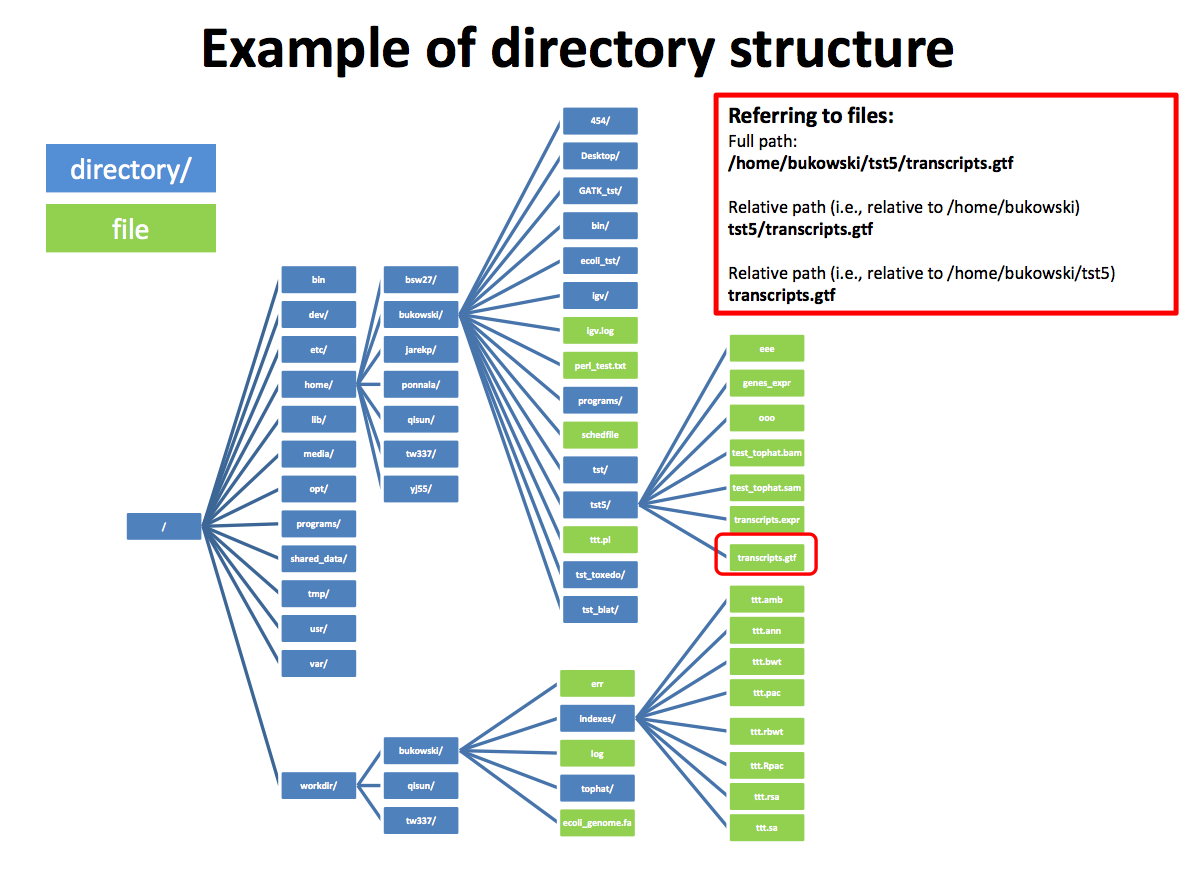
Simple commands
Comments are declared by # symbol. The computer ignores these lines, and are a good practice for documentation
user40
#show your location/ Workspace
/global/home/user40
# cd : Change Directory to default.
[user40@hpc ~]$ pwd
/global/home/user40
# cd .. : Change Directory to parent (denoted by the two dots).
[user40@hpc home]$ pwd
/global/home
[user40@hpc global]$ pwd
/global
[user40@hpc /]$ pwd
/
[user40@hpc ~]$ pwd
/global/home/user40
[user40@hpc global]$ pwd
/global
[user40@hpc global]$ cd ~
[user40@hpc ~]$ pwd
/global/home/user40
Paths can be absolute as in
Looking at files and directories
fasta_files galaxy-dist galaxy-python
[user40@hpc ~]$ ls /global/
courses galaxy home scratch software
[user40@hpc ~]$ ls ..
[user40@hpc ~]$ ls -l
total 8
drwxr-xr-x 2 user40 cgrl 4096 Mar 11 14:45 fasta_files
drwxr-xr-x 21 user40 cgrl 4096 Feb 26 10:13 galaxy-dist
drwxr-xr-x 2 user40 cgrl 19 Jan 16 12:05 galaxy-python
total 88
drwx------ 11 user40 cgrl 4096 Mar 11 14:04 .
drwxr-xr-x 70 root root 4096 Feb 19 18:28 ..
-rw-r--r-- 1 user40 cgrl 8891 Feb 19 15:58 .RData
-rw------- 1 user40 cgrl 154 Feb 19 15:58 .Rhistory
-rw------- 1 user40 cgrl 21333 Mar 18 18:24 .bash_history
-rw-r--r-- 1 user40 cgrl 33 Jan 7 09:57 .bash_logout
-rw-r--r-- 1 user40 cgrl 203 Jan 28 12:52 .bash_profile
-rw-r--r-- 1 user40 cgrl 124 Jan 7 09:57 .bashrc
-rw-r--r-- 1 user40 cgrl 515 Jan 7 09:57 .emacs
drwx------ 2 user40 cgrl 6 Feb 19 16:22 .gconf
drwx------ 2 user40 cgrl 24 Feb 19 16:22 .gconfd
drwx------ 3 user40 cgrl 16 Feb 19 17:12 .local
-rw------- 1 user40 cgrl 6 Jan 29 15:06 .psql_history
drwxr-xr-x 7 user40 cgrl 4096 Jan 7 12:25 .python-eggs
drwx------ 2 user40 cgrl 24 Jan 28 10:35 .ssh
drwxr-xr-x 3 root root 61 Feb 19 17:51 .subversion
-rw------- 1 user40 cgrl 7263 Feb 19 17:38 .viminfo
drwxr-xr-x 2 user40 cgrl 4096 Mar 11 14:45 fasta_files
drwxr-xr-x 21 user40 cgrl 4096 Feb 26 10:13 galaxy-dist
drwxr-xr-x 2 user40 cgrl 19 Jan 16 12:05 galaxy-python
total 8
drwxr-xr-x 2 user40 cgrl 4096 Mar 11 14:45 fasta_files
drwxr-xr-x 21 user40 cgrl 4096 Feb 26 10:13 galaxy-dist
drwxr-xr-x 2 user40 cgrl 19 Jan 16 12:05 galaxy-python
./ .RData.bash_history .bash_profile .emacs .gconfd/ .psql_history.ssh/ .viminfo galaxy-dist/
../ .Rhistory.bash_logout .bashrc .gconf/ .local/ .python-eggs/.subversion/ fasta_files/ galaxy-python/
fasta_files/:
GA_repeats.txtSGD_features.tab cerevisiae_genome.fasta fasta_dir md5sum.txt
fasta_files/fasta_dir:
chrI.fa chrIII.fa chrIX.fa chrV.fa chrVII.fa chrX.fa chrXII.fa chrXIV.fa chrXVI.fa
chrII.fa chrIV.fa chrM.fa chrVI.fa chrVIII.fa chrXI.fa chrXIII.fa chrXV.fa
You can use tab key to autocomplete after you type the first few letters of a file or a directory or even a command. Hitting tab twice will list all the possibilities. Beware this might fill up your screen very fast if there are a lot of possibilities.
You can also use the up and down arrow keys to cycled through your previously typed commands. This saves you a lot of re-typing and reduces errors.
Another useful command is the history command. This prints to screen all your previous commands and their line numbers.
1037 ls
1038 cd ..
1039 clear
1040 ls -R fasta_files/
1041 ls -Sr
1042 ls -S
1043 ls -s
1044 ls ..
1045 pwd
1046 ls /global/
1047 ls -l /global/
1048 clear
1049 history
You can re run any command in the history by using the command's line number in history
ls /global/
courses galaxy home scratch software
Creating files and directories
[user40@hpc ~]$ ls -l
total 4
drwxr-xr-x 3 user40 cgrl 113 Mar 18 18:35 fasta_files
drwxr-xr-x 21 user40 cgrl 4096 Feb 26 10:13 galaxy-dist
drwxr-xr-x 2 user40 cgrl 19 Jan 16 12:05 galaxy-python
drwxr-xr-x 2 user40 cgrl 6 Mar 18 18:54 unix_playground
[user40@hpc unix_playground]$ touch my_first_file
[user40@hpc unix_playground]$ ls -l
total 0
-rw-r--r-- 1 user40 cgrl 0 Mar 18 18:55 my_first_file
[user40@hpc unix_playground]$ ls -l
total 0
-rw-r--r-- 1 user40 cgrl 0 Mar 18 18:55 My_First_file
-rw-r--r-- 1 user40 cgrl 0 Mar 18 18:55 my_first_file
[user40@hpc unix_playground]$ ls -l
total 0
-rw-r--r-- 1 user40 cgrl 0 Mar 18 18:55 My_First_file
-rw-r--r-- 1 user40 cgrl 0 Mar 18 18:56 my_first_file
Notice how touch recreated a new file with an updated time stamp. This is the behavior if file already exists, so be careful not to overwrite an existing file.
A few important things to learn from this:
- file and directory names are case sensitive in linux.
- Always name your files and directories something short and informative and use extensions (such as .fasta or .fastq to let you know what kind of file it is).
- Avoid using spaces in your file and directory names. We will see later why.
Modifying files and directories
[user40@hpc unix_playground]$ ls -l
total 0
-rw-r--r-- 1 user40 cgrl 0 Mar 18 18:56 my_first_file
[user40@hpc unix_playground]$ ls -l
total 0
-rw-r--r-- 1 user40 cgrl 0 Mar 18 18:56 my_first_file
drwxr-xr-x 2 user40 cgrl 6 Mar 18 19:05 temp1
drwxr-xr-x 2 user40 cgrl 6 Mar 18 19:05 temp2
[user40@hpc unix_playground]$ rmdir temp1/
total 0
-rw-r--r-- 1 user40 cgrl 0 Mar 18 18:56 my_first_file
drwxr-xr-x 2 user40 cgrl 6 Mar 18 19:05 temp2
[user40@hpc temp2]$ touch test.txt
[user40@hpc temp2]$ cd ..
[user40@hpc unix_playground]$ rmdir temp2/
rmdir: temp2/: Directory not empty
[user40@hpc unix_playground]$ ls -l
total 0
-rw-r--r-- 1 user40 cgrl 0 Mar 18 18:56 my_first_file
[user40@hpc unix_playground]$ ls -l
total 0
-rw-r--r-- 1 user40 cgrl 0 Mar 18 18:56 my_first_move.txt
[user40@hpc unix_playground]$ mv my_first_move.txt move_dir/
[user40@hpc unix_playground]$ ls move_dir/
my_first_move.txt
[user40@hpc unix_playground]$ ls -l
total 0
drwxr-xr-x 2 user40 cgrl 30 Mar 18 19:08 move_dir
-rw-r--r-- 1 user40 cgrl 0 Mar 18 19:09 my_first_copy.txt
Be careful with
Adding text to a file
Copy the following and paste into the txt file and then ctrl+o to write and ctrl+x to exit
>seq1 This is the description of my first sequence.
AGTACGTAGTAGCTGCTGCTACGTGCGCTAGCTAGTACGTCAA
TCGTACGTCGACTGATCGTAGCTACGTCGTACGTAGGTACGTT
CGACGTAGATGCTAGCTGACTCGATGC
>seq2 This is a description of my second sequence.
CGATCGATCGTACGTCGACTGATCGTAGCTACGTCGTACGTAG
GTACGTGATCGTACGTCGACTGATCGTAGCTATCGTAGCTACC
CATCGTCAGTTACTGCATGCTCG
>seq3 and so on...
GATCGTACGTGTAGCTGCTGTAGCTGATCGTACGGTAGCAGCT
CGTCGACTGATCGTAGCTGATCGTACGTCGACTGATCGTAGCT
AGTACGTAGTAGCTGTAGCTGATCGTACGTAGCTGATCGTACG
>seq4
CGATCGATCGTACGTCGACTGATCGTAGCTACGTCGTACGTAG
GTACGTGATCGTACGTCGACTGATCGTAGCTATCGTAGCTACC
GTAGATGCTAGCTGACTCGAT
Peeking inside text based files
>seq1 This is the description of my first sequence.
AGTACGTAGTAGCTGCTGCTACGTGCGCTAGCTAGTACGTCAA
TCGTACGTCGACTGATCGTAGCTACGTCGTACGTAGGTACGTT
CGACGTAGATGCTAGCTGACTCGATGC
>seq2 This is a description of my second sequence.
CGATCGATCGTACGTCGACTGATCGTAGCTACGTCGTACGTAG
GTACGTGATCGTACGTCGACTGATCGTAGCTATCGTAGCTACC
CATCGTCAGTTACTGCATGCTCG
>seq3 and so on...
GATCGTACGTGTAGCTGCTGTAGCTGATCGTACGGTAGCAGCT
CGTCGACTGATCGTAGCTGATCGTACGTCGACTGATCGTAGCT
AGTACGTAGTAGCTGTAGCTGATCGTACGTAGCTGATCGTACG
>seq4
CGATCGATCGTACGTCGACTGATCGTAGCTACGTCGTACGTAG
GTACGTGATCGTACGTCGACTGATCGTAGCTATCGTAGCTACC
GTAGATGCTAGCTGACTCGAT
>seq1 This is the description of my first sequence.
AGTACGTAGTAGCTGCTGCTACGTGCGCTAGCTAGTACGTCAA
TCGTACGTCGACTGATCGTAGCTACGTCGTACGTAGGTACGTT
CGACGTAGATGCTAGCTGACTCGATGC
>seq2 This is a description of my second sequence.
CGATCGATCGTACGTCGACTGATCGTAGCTACGTCGTACGTAG
GTACGTGATCGTACGTCGACTGATCGTAGCTATCGTAGCTACC
CATCGTCAGTTACTGCATGCTCG
>seq3 and so on...
GATCGTACGTGTAGCTGCTGTAGCTGATCGTACGGTAGCAGCT
CGTCGACTGATCGTAGCTGATCGTACGTCGACTGATCGTAGCT
AGTACGTAGTAGCTGTAGCTGATCGTACGTAGCTGATCGTACG
>seq4
CGATCGATCGTACGTCGACTGATCGTAGCTACGTCGTACGTAG
GTACGTGATCGTACGTCGACTGATCGTAGCTATCGTAGCTACC
GTAGATGCTAGCTGACTCGAT
Press q to exit out of this
>seq1 This is the description of my first sequence. AGTACGTAGTAGCTGCTGCTACGTGCGCTAGCTAGTACGTCAA TCGTACGTCGACTGATCGTAGCTACGTCGTACGTAGGTACGTT CGACGTAGATGCTAGCTGACTCGATGC >seq2 This is a description of my second sequence. CGATCGATCGTACGTCGACTGATCGTAGCTACGTCGTACGTAG GTACGTGATCGTACGTCGACTGATCGTAGCTATCGTAGCTACC CATCGTCAGTTACTGCATGCTCG >seq3 and so on... GATCGTACGTGTAGCTGCTGTAGCTGATCGTACGGTAGCAGCT
GTACGTGATCGTACGTCGACTGATCGTAGCTATCGTAGCTACC CATCGTCAGTTACTGCATGCTCG >seq3 and so on... GATCGTACGTGTAGCTGCTGTAGCTGATCGTACGGTAGCAGCT CGTCGACTGATCGTAGCTGATCGTACGTCGACTGATCGTAGCT AGTACGTAGTAGCTGTAGCTGATCGTACGTAGCTGATCGTACG >seq4 CGATCGATCGTACGTCGACTGATCGTAGCTACGTCGTACGTAG GTACGTGATCGTACGTCGACTGATCGTAGCTATCGTAGCTACC GTAGATGCTAGCTGACTCGAT
>seq1 This is the description of my first sequence. AGTACGTAGTAGCTGCTGCTACGTGCGCTAGCTAGTACGTCAA TCGTACGTCGACTGATCGTAGCTACGTCGTACGTAGGTACGTT
CGATCGATCGTACGTCGACTGATCGTAGCTACGTCGTACGTAG GTACGTGATCGTACGTCGACTGATCGTAGCTATCGTAGCTACC GTAGATGCTAGCTGACTCGAT
If you need help about any command or its options use
This is very useful when you learning command line.
A few more handy utilities
This is my second file! I am getting good at this.
>seq1 This is the description of my first sequence.
AGTACGTAGTAGCTGCTGCTACGTGCGCTAGCTAGTACGTCAA
TCGTACGTCGACTGATCGTAGCTACGTCGTACGTAGGTACGTT
CGACGTAGATGCTAGCTGACTCGATGC
>seq2 This is a description of my second sequence.
CGATCGATCGTACGTCGACTGATCGTAGCTACGTCGTACGTAG
GTACGTGATCGTACGTCGACTGATCGTAGCTATCGTAGCTACC
CATCGTCAGTTACTGCATGCTCG
>seq3 and so on...
GATCGTACGTGTAGCTGCTGTAGCTGATCGTACGGTAGCAGCT
CGTCGACTGATCGTAGCTGATCGTACGTCGACTGATCGTAGCT
AGTACGTAGTAGCTGTAGCTGATCGTACGTAGCTGATCGTACG
>seq4
CGATCGATCGTACGTCGACTGATCGTAGCTACGTCGTACGTAG
GTACGTGATCGTACGTCGACTGATCGTAGCTATCGTAGCTACC
GTAGATGCTAGCTGACTCGAT
16 35 598 my_first_fasta.fasta
16 my_first_fasta.fasta
598 my_first_fasta.fasta
Cut can be used to extract columns from a delimited file.
Can you think on how would you extract columns if the file was a csv??
Sort can be used to sort based on any given column
LOC_001608XLOC_001608ZYG11Bchr1:53192130-53293013MockOGTOK05790.27infnan0.136850.42102no XLOC_000409XLOC_000409ZYG11Bchr1:53192130-53293013MockOGTOK11.941513.21850.1465790.1444550.82990.998471no XLOC_031804XLOC_031804ZYXchr7:143078359-143220540MockOGTOK5.088096.067640.254010.1167770.716950.996395no XLOC_014317XLOC_014317ZZEF1chr17:3907738-4046253MockOGTOK2.743513.341760.2845830.1992790.658150.977286no XLOC_001676XLOC_001676ZZZ3chr1:78030189-78148343MockOGTOK42.826824.3581-0.814114-0.6450210.29120.682889no test_idgene_idgenelocussample_1sample_2statusvalue_1value_2log2(fold_change)test_statp_valueq_valuesignificant
How would you sort this in a reverse order? UseSort a file by column 4 with -n option and without -n option, what is the difference?
What does the
Redirection and piping
Let us get the first 100 lines of gene_exp.diff and save it to a new file. How do we do this?[user40@hpc unix_playground]$ head -100 gene_exp.diff > gene_exp_first_100_lines.diff
[user40@hpc unix_playground]$ wc -l gene_exp.diff gene_exp_first_100_lines.diff
37605 gene_exp.diff 100 gene_exp_first_100_lines.diff 37705 total
This > sign is used to redirect the output of head into a new file called gene_exp_first_100_lines.diff This will overwrite whatever is in the recipient file name. To append use >> sign
[user40@hpc unix_playground]$ head gene_exp_first_100_lines.diff
I am adding a new line
[user40@hpc unix_playground]$ head -100 gene_exp.diff > gene_exp_first_100_lines.diff
[user40@hpc unix_playground]$ echo "I am adding a new line" >> gene_exp_first_100_lines.diff
[user40@hpc unix_playground]$ tail -5 gene_exp_first_100_lines.diff
XLOC_000096XLOC_000096PLOD1chr1:11994723-12035599MockOGTOK4.9559657.11413.526610.8052650.082050.288638no XLOC_000097XLOC_000097MFN2chr1:12040237-12073572MockOGTOK22.837323.35650.03243250.01837010.97060.998471no XLOC_000098XLOC_000098MIIPchr1:12079298-12092106MockOGTOK10.270610.80530.07321580.06297720.8970.998471no XLOC_000099XLOC_000099TNFRSF8chr1:12123433-12204264MockOGTOK0.1032310.4866612.237040.6853430.02570.224628no I am adding a new line
Let's say we want to get the gene name column (column 3) and then sort the output. How would we do this??
This is where we use pipes, pipes take the output of one command and use it as input for the command after the pipe. Useful for chaining commands which have one output and accept one input
How would you use pipes to give me a list of only unique gene names in this file?
Wildcards
A lot of commands can take multiple inputs, like touch, mkdir, rm, cp, mv, wc etc So if we are able to pass multiple files (or dir) to these commands, they will work on all of them. We could do this file by file or we could use an expression to match these multiple files. This is where wildcards come in. The following are a few wildcards we will talk about. is a wildcard that can match zero or more characters of any kind
? is a wildcard that can match 1 occurrence of any character
[ABC] can match for any of the characters occurring inside them, A or B or C
! negates match expressions [!ABC], any character except A or B or C
{A,B,C} expands all the elements within the braces
{1..9} expands a range of elements within the braces
[user40@hpc unix_playground]$ cd wildcards/
[user40@hpc wildcards]$ touch file{1..6}.txt
[user40@hpc wildcards]$ ls
file1.txt file2.txt file3.txt file4.txt file5.txt file6.txt
[user40@hpc wildcards]$ ls
file1.txt file2.txt file3.txt file4.txt file5.txt file6.txt file_a.log file_b.log file_c.log file_d.log file_e.log
file1.txt file2.txt file3.txt file4.txt file5.txt file6.txt
file1.txt file2.txt file3.txt file4.txt file5.txt file6.txt file_a.log file_b.log file_c.log file_d.log file_e.log
????
[user40@hpc wildcards]$ ls file?.*
????
???
[user40@hpc wildcards]$ ls
???
[user40@hpc wildcards]$ ls
[!46].txt file4.txt file6.txt file_a.log file_b.log file_c.log file_d.log file_e.log
How do you remove this file [!46].txt? Hint: You would have to escape the [ and ! and ] to make them not special.
Remove all the files in this directory. And then navigate out of the directory and remove the directory named wildcards
Searching for files and their contents
Look upFind is useful for finding file names that match certain criteria
unix_playground/my_first_fasta.fasta
unix_playground/second_file.txt
unix_playground/gene_exp.diff
unix_playground/gene_exp_first_100_lines.diff
unix_playground/
unix_playground/second_file.txt
unix_playground/my_first_fasta.fasta
???
16 unix_playground/my_first_fasta.fasta
1 unix_playground/second_file.txt
37605 unix_playground/gene_exp.diff
101 unix_playground/gene_exp_first_100_lines.diff
37723 total
Here we are using the output of find by placing its output in parenthesis with a $ and passing it to wc. This is another way of passing arguments to commands.
>seq1 This is the description of my first sequence.
>seq2 This is a description of my second sequence.
>seq3 and so on...
>seq4
1:>seq1 This is the description of my first sequence. 5:>seq2 This is a description of my second sequence. 9:>seq3 and so on... 13:>seq4
1:>
5:>
9:>
13:>
2:AGTACGTAGTAGCTGCTGCTACGTGCGCTAGCTAGTACGTCAA
3:TCGTACGTCGACTGATCGTAGCTACGTCGTACGTAGGTACGTT
4:CGACGTAGATGCTAGCTGACTCGATGC
6:CGATCGATCGTACGTCGACTGATCGTAGCTACGTCGTACGTAG
7:GTACGTGATCGTACGTCGACTGATCGTAGCTATCGTAGCTACC
8:CATCGTCAGTTACTGCATGCTCG
10:GATCGTACGTGTAGCTGCTGTAGCTGATCGTACGGTAGCAGCT
11:CGTCGACTGATCGTAGCTGATCGTACGTCGACTGATCGTAGCT
12:AGTACGTAGTAGCTGTAGCTGATCGTACGTAGCTGATCGTACG
14:CGATCGATCGTACGTCGACTGATCGTAGCTACGTCGTACGTAG
15:GTACGTGATCGTACGTCGACTGATCGTAGCTATCGTAGCTACC
16:GTAGATGCTAGCTGACTCGAT
77:XLOC_000076XLOC_000076TMEM201chr1:9648931-9674935MockOGTOK1.615331.53688-0.0718273-0.05297790.900450.998471no
How would you get the header file in the gene_exp.diff using grep?
You can use egrep which is a more powerful form of grep and you can specify wildcards and regular expression to search for patterns.
as an example we could use egrep to find fasta files which end in 'T' for example
3:TCGTACGTCGACTGATCGTAGCTACGTCGTACGTAGGTACGTT 10:GATCGTACGTGTAGCTGCTGTAGCTGATCGTACGGTAGCAGCT 11:CGTCGACTGATCGTAGCTGATCGTACGTCGACTGATCGTAGCT 16:GTAGATGCTAGCTGACTCGAT
We could find entries that begin with C and end with T
11:CGTCGACTGATCGTAGCTGATCGTACGTCGACTGATCGTAGCT
Hands-on Exercises
Exercise 1
Make a new directory called fasta_files and download files from ftp://hgdownload.cse.ucsc.edu/goldenPath/sacCer3/chromosomes/* usingThen do the following:
a) Unzip the downloaded file (HINT: use gunzip, look at its documentation for help)
b) Change into your fasta_files directory, and unzip the remainder of the fasta files using only a one-line command.
c) Combine the sequence files to make a single whole genome file called "cerevisiae_genome.fasta"
d) Count the chromosomes in the whole genome file using commands from the lecture. (HINT: Each of the original FASTA files contains a single chromosome).
e) Get size of total genome. (HINT: The size of the genome can be determined by counting the number of bases).
Exercise 2
- Get the list of cerevisiae chromosome features from this address: http://downloads.yeastgenome.org/curation/chromosomal_feature/SGD_features.tab
- Next, count the total number of ORFs in the features file.
- Now count the ORFs listed as Verified, and separately count those listed as Dubious. If you do the math and add up these two groups, why don't they make up all the ORF matches fromgrep?
- Use the cut utility with grep to count the number of ORFs more accurately.
- Use a combination of the the commands cut, sort, and uniq generate a list of all the genomic features (column 2) in this file. Don't forget that you can use the man pages to learn how to use new tools.
Columns within SGD_features.tab:
1. Primary Stanford Gene Database ID (SGDID) (mandatory)
2. Feature type (mandatory)
3. Feature qualifier (optional)
4. Feature name (optional)
5. Standard gene name (optional)
6. Alias (optional, multiples separated by |)
7. Parent feature name (optional)
8. Secondary SGDID (optional, multiples separated by |)
9. Chromosome (optional)
10. Start_coordinate (optional)
11. Stop_coordinate (optional)
12. Strand (optional)
13. Genetic position (optional)
14. Coordinate version (optional)
15. Sequence version (optional)
16. Description (optional)
Exercise 3
Is there a way to tell grep to ONLY print the part of the line that matches the pattern you give it?Use your new found knowledge of grep and egrep, combined with some of the other unix utilities you learned about earlier, to produce a table that lists every length of GA microsatellite repeat in the yeast genome, along with the number of occurrences of each. You can do this with a single command, piping output between the different unix utilities.
Example:
5000 GA
3000 GAGA
1000 GAGAGA
100 GAGAGAGA
10 GAGAGAGAGA
...
...
HINT #1: You will need to use an extra argument with the uniq command
HINT #2: Regular expressions provide the flexibility to match patterns rather than a specific string. You can use special characters to construct a regular expression, for example, the plus sign means match one or more times, so the pattern 'H+' will match the H in HAT but also the HHHHHHHH in AHHHHHHHH!!!!!!! The period meansmatch any character so, from the previous example, 'H.+' would match all of HAT as well as HHHHHHHH!!!!!!!. You can also group characters together with parentheses. For example, '(I..)+' would match ISSISSIPP in MISSISSIPPI. Remember to enclose your pattern in 'tick marks' just like with echo.
CAVEAT: Assume that it is safe to ignore the fact that a repeat could possibly span multiple lines in your FASTA file
Save your table as a new file. Open the file in emacs and add column headers and/or anything else you can think of to make it pretty. Save the file and exit.
Shell Scripting
We can write scripts that run in the bash shell to automate a lot of our commands. We can use this to run a set of commands repeatedly over a bunch of different files. We could use wild cards but not always. For example to get lines 15-20 of every fasta file in fasta_files directory.CATTTATATACACTTATGTCAATATTACAGAAAAATCCCCACAAAAATCA CCTAAACATAAAAATATTCTACTTTTCAACAATAATACATAAACATATTG GCTTGTGGTAGCAACACTATCATGGTATCACTAACGTAAAAGTTCCTCAA TATTGCAATTTGCTTGAACGGATGCTATTTCAGAATATTTCGTACTTACA CAGGCCATACATTAGAATAATATGTCACATCACTGTCGTAACACTCTTTA
Lets make a bash script to do this for all the files, building our code one step at a timehead -20 chrI.fa | tail -5
into the file and write and exit.
CATTTATATACACTTATGTCAATATTACAGAAAAATCCCCACAAAAATCA
CCTAAACATAAAAATATTCTACTTTTCAACAATAATACATAAACATATTG
GCTTGTGGTAGCAACACTATCATGGTATCACTAACGTAAAAGTTCCTCAA
TATTGCAATTTGCTTGAACGGATGCTATTTCAGAATATTTCGTACTTACA
CAGGCCATACATTAGAATAATATGTCACATCACTGTCGTAACACTCTTTA
Change your subset.sh to the following
CCGAAATCCAAGAAACTGTAAGACATTCATATTCTCGGAAGTATTGGGAA
ATTGTGCTTTCAGTTTCTTTCTCTCTAGCAAAACCATTTGACTCCCTTTC
CGCTTATACGACTCTTTGTTAATGTCGGTGACTGGATGGAATCTATTATC
CTCAGCATTGCCATCTTTATTGGCGTCCTCCTTGGCACTAGCGTTGGTGC
TGGCAGTGGTAGTAGCATTAGTCCTGACGTTGATGCTGGCAGTGGTAGTC
head -${2} $1 | tail -${3}
Now when you run it again we can give it arguments for subsetting any number of lines.
TTCTTCCAAATTTTATGAACATGCCCTAAAGGCACCTCGGATTTCTCCTT
GATTAGATTAAACATCCGTGTTGGATAGCTGGATAGACCTCTGCTGAGAT
CTTCCGACCGTTTGAGCTCGTTGATGTCCATCGACTTCTTGGCCAGTCCC
GTAAGCCCAATACGCTGCAACGCAGCATCAGCTACAGCCTCAGGGGCTGT
GCCGCTCAAAAAGATTGCTTTCTCAAAAGCGTCAAAATCAAGGTTAGTTA
TGCCCCCAAATTGCGACTGCCGGTAGACCTCCGTTTCAAAGTTGTGAAAC
TCATCTACAATGAGGTAACCCAATTTTACGTTGTTGGTCCTAAAGGTGCA
CTCAACAATATTCTCCCACGCAGCTATCCTGTCTGTGAAATTAGTGCTAG
CAAGATCATCGTAGATCCCCACGTATAAATCAGTAACGCCATCGTAACCT
TCTTCAATAAAGTTTCTTACAGGGGCCACATTCAAGCAACCGCGTCGGCC
Change subset.sh to this
do
echo "lines 15-20 of" $fasta
head -20 $fasta | tail -5
done
[user40@hpc fasta_files]$ bash subset.sh chr*
This is an example of for loops in a shell script, we are looping over each element of $* and assigning to a variable fasta and we are accessing that fasta file through $fasta and getting lines 15-20 and printing out. We could pipe the output and save it. There are other loops you can run in bash script as well. For example you could run Let us make it such that we don't have to type bash every time.
Add the following lines to the top of your shell script
This tells the terminal to use bash to execute the script. But now we also need to make the script executable
-rw-r--r-- 1 user40 cgrl 79 Mar 18 21:43 subset.sh
Notice how the permissions are not setup with an x to execute this script. So we will change permissions to allow execution[user40@hpc fasta_files]$ ls -l subset.sh
-rwxr-xr-x 1 user40 cgrl 79 Mar 18 21:43 subset.sh
We still need to type ./ because terminal is not searching the current directory for executables, for example when you type ls or cd or mv these are actually present in the PATH.
/bin/ls
So /bin/ must be in the path because we are able to type ls. To check this we could do/usr/kerberos/bin:/usr/local/bin:/bin:/usr/bin:/opt/dell/srvadmin/bin:/global/home/user40/bin
Indeed it is. So to get our subset.sh to run like ls or any other commands that are in the PATH, we need to add our current directory to the PATH, how do we do this?[user40@hpc fasta_files]$ pwd
/global/home/user40/unix_playground/fasta_files
/global/home/user40/unix_playground/fasta_files:/usr/kerberos/bin:/usr/local/bin:/bin:/usr/bin:/opt/dell/srvadmin/bin:/global/home/user40/bin
See how my pwd is now in the path so I can start typing sub and use tab to file in
subset.sh
Exercise bash scripting
Rename all the fasta files to so chr is now chromosome for example chrV.fa is now chromosomeV.faHINT:
${variable/search_tem/replace_term}
will return a variable with search_term replaced with replace_term