BLAST database search
BLAST: Basic Local Alignment Search Tool.
BLAST program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance of matches.Establishing links between observed sequence variation and gene function is a major challenge when analyzing transcriptome data from non-model organisms. Here, the Basic Local Alignment Search Tool (BLAST) is used to compare your de novo assembled contigs to sequence databases in order to annotate them with similarity to known genes/proteins/functions. BLAST is a toolkit developed by the National Center for Biotechnology Information (NCBI), the US-based organization responsible for archiving and databasing the world's genetic sequence information.
BLAST homepage
A note on E-values
To determine whether matches to the databases are "significant", we use a threshold E-value. The E-value describes the number of hits one can expect to see by chance when searching a database of a particular size. The lower the E-value, the more "significant" a match to a database sequence is (i.e. there is a smaller probability of finding a match just by chance).
However, the quality of a match also depends on the length of the alignment and the percentage similarity, so these statistics may also be considered when evaluating the significance of a match.
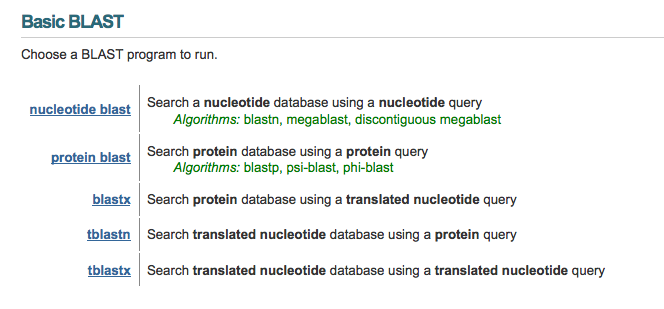
BLAST programs
Guide how to do a Protein sequence search
Program: BLASTP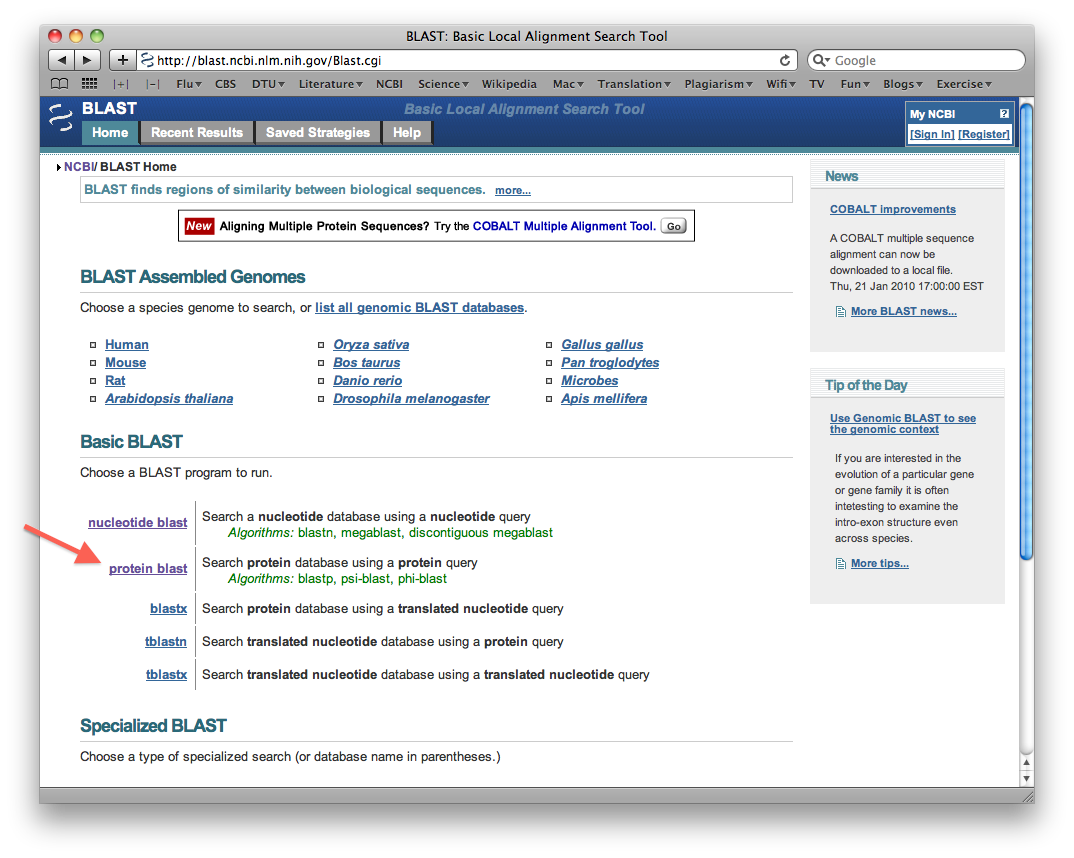
Enter a random protein sequence that is 40 amino acids long in the sequence window. Make sure to write the sequence in fasta format (recall: name line begins with ">").
>unknown_proteinYou can copy paste this sequence
MATGSRTSLLLAFGLLCLPWLQEGSAFPTIPLSRLFDNAMLRAHRLHQLAFDTYQEFE EAYIPKEQKYSFLQNPQTSLCFSESIPTPSNREETQQKSNLELLRISLLLIQSWLEPV QFLRSVFANSLVYGASDSNVYDLLKDLEEGIQTLMGRLEDGSPRTGQIFKQTYSKFDT NSHNDDALLKNYGLLYCFRKDMDKVETFLRIVQCRSVEGSCGF
Select "Show results in a new window", and then search the database by clicking the big BLAST button:
Program: BLASTP
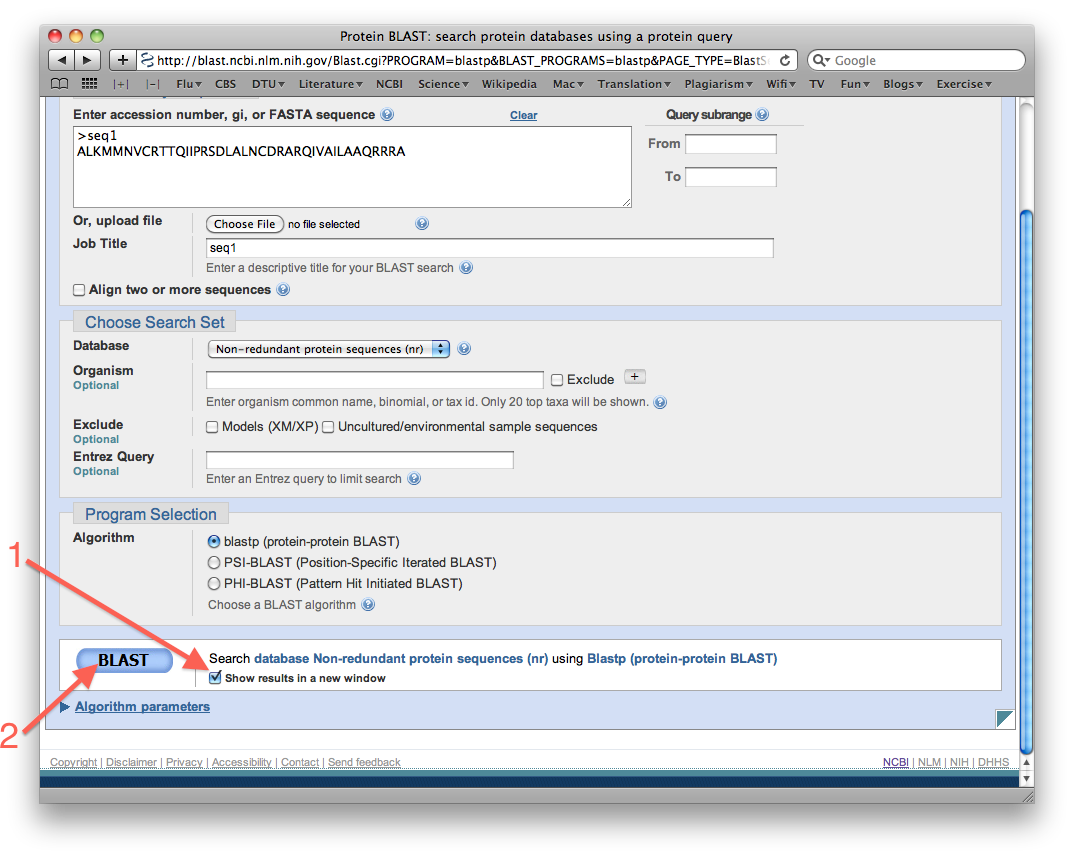
Under algorith parameters, at the bottom of hte page, you can adjust optional parameters, such as e-value, maximum target sequences, etc.
- Based on the hits can you predict the function of this unknown protein sequence?
Predicting the function of un-characterised proteins by finding similar, known proteins in the database, is probably the single most important bioinformatics method!
BLAST FOR BEGINNERS
Click here for a quick guide and introduction to BLAST [courtesy of Sandra Porter; digitalworldbiology]
Command line BLAST
While The NCBI web-based BLAST is graphical, i.e. gives results in intuitive, color-coded grapgics according to the score of the alignment, its over reliance on internet is a setback. Also, it is slow, and tedious while processing many sequences / Contigs.- Download the sequences for which you will search your query against (This may be at kingdom, phylum, class, order, family, genus, or species level)
- Format the sequences as database
- Geting help with the BLAST programs It is not possible to cover each and every detail of arguments used for each program. To get an overview of how this program works and which arguments and parameter values you can input, take a look at the help page, which you open in Terminal by typing:
- Running a query search: An example command to search a contig against a protein database is shown below:
The –in parameter specifies the name of the .fasta file that contains the sequences you want to format dopwnloaded above;
-dbtype specifies whether you are dealing with a nucleotide or protein database ; and
-out specifies the name you want to use for your formatted database.
blastx -query YOURASSEMBLY.fasta -db DBNAME -out YOURASSEMBLY_BLASTX2DBNAME -outfmt 6 -evalue 0.0001 -gapopen 11 -gapextend 1 -word_size 3 -matrix BLOSUM62 -num_threads 4 The -query is the input file name, e.g. testfile_assembly.fasta;
-db is the name you gave to your formatted BLAST database above, e.g. NR (note that you should not specify the extensions for the formatted database file names (.phr, .pin and .psq) only the given name of the file);
-out is the name you want for your output file, e.g. testfile_blastx2NR;
-outfmt specifies the format for how the program outputs the results;
–evalue is the expectation value (E) threshold for saving hits- your choice should depend on how stringently you want to exclude potential chance matches);
-gapopen, –gapextend, word_size, and –matrix specify parameters for the alignment algorithm
(see http://www.ncbi.nlm.nih.gov/BLAST/blastcgihelp.shtml for details);
-num_threads is the number of threads the BLAST search should use (depends on how many processors/cores your computer runs on).
For many of the parameters, we use the default values from the NCBI web-based BLAST searches. However, to keep track of analysis settings, we have found it helpful to specify these parameters with every search as different versions of the stand-alone BLAST toolkit may have different default parameters. Feel free to modify these, but the parameters specified here should provide a good starting point.