This chapter is about sequence similarity. Let us start with a warning: there is no unique, precise, or universally applicable notion of similarity. An alignment is an arrangement of two sequences which shows where the two sequences are similar, and where they differ. An optimal alignment, of course, is one that exhibits the most significant similarities, and the least differences. Broadly, there are three categories of methods for sequence comparison.
unix % dottup
DNA sequence dot plot
Input sequence: embl:xl23808
Second sequence: embl:xlrhodop
Word size [4]: 10
Graph type [x11]:
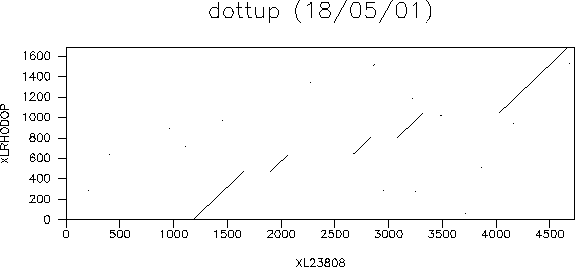
A window will pop up on your screen that should look something like this:
The diagonal lines represent areas where the two sequences align well. You can see that there are five clear diagonals. You will remember that we are aligning genomic and cDNA - these five diagonals represent the five exons of the gene! If you look at the original EMBL entry for the genomic sequence using SRS, you will see that the annotated entry says that there are five exons in this gene. So our results are in agreement.
The settings we have used for this example are those that give the best results. dottup looks for exact matches between sequences. As we expect the exon regions from the genomic sequence to exactly match the cDNA sequence we can use longer word lengths as we should still get exact matches. This gives a very clean plot. If you were to match the cDNA sequence against that of a related sequence, e.g. the rhodopsin from mouse (embl:m55171) then you wouldn't expect long exact matches so should use a shorter word length.
Repeat the previous example comparing the frog rhodopsin cDNA against the mouse genomic DNA:
unix % dottup embl:m55171 embl:xlrhodop
DNA sequence dot plot
Word size [4]: 10
Graph type [x11]:
Repeat this for both comparisons using different word sizes. What do you notice?
Which word sizes give the clearest plot?
Why are the diagonals in the first and last exons not so clear? (hint: look back at the results for showfeat)
The dotplot doesn't give us any detailed sequence information. For
this, we need to use different programs. The algorithms we will be
using are more rigorous than those used for searching databases; so
even if you have retrieved a sequence from a database using something
like BLAST, it will be well worth your while performing a careful
pairwise alignment afterwards. The basic idea behind the sequence
alignment programs is to align the two sequences in such a way as to
produce the highest score - a scoring matrix is used to add points to
the score for each match and subtract them for each mismatch. The
matrices used for nucleic acid alignments tend to involve fairly
simple match/mismatch scoring schemes, while the matrices commonly
used for scoring protein alignments are more complex, with scores
designed to reflect similarity between the different amino acids
rather than simply scoring identities. Over time various mutations
occur in sequences; the scoring matrices attempt to cope with
mutations, but insertions and deletions require some extra parameters
to allow the introduction of gaps in the alignment. There are
penalties both for the creation of gaps and for the extension of
existing ones; the default gap parameters given in alignment programs
have been found to be empirically correct with test sequences but you
should experiment with different gap penalties.
A global alignment is one that compares the two sequences over their entire lengths, and is appropriate for comparing sequences that are expected to share similarity over the whole length. The alignment maximises regions of similarity and minimises gaps using the scoring matrices and gap parameters provided to the program. The EMBOSS program needle is an implementation of the Needleman-Wunsch [] algorithm for global alignment; the computation is rigorous and needle can be time consuming to run if the sequences are long.
unix % needle
Needleman-Wunsch global alignment.
Input sequence:embl:xlrhodop
Second sequence:embl:xl23808
Gap opening penalty [10.0]:
Gap extension penalty [0.5]:
Output file [xlrhodop.needle]:
unix % more xlrhodop.needle
Global: XLRHODOP vs XL23808 Score: 7471.00 XLRHODOP XL23808 1 cgtaactaggaccccaggtcgacacgacaccttccctttcccagt 45 XLRHODOP XL23808 46 tatttcccctgtagacgttagaaggggaaggggtgtacttatgtc 90 XLRHODOP XL23808 91 acgacgaactacgtccttgactacttagggccagagagacgaggt 135
XLRHODOP 1 ggtagaacagcttcagttgggatcacaggcttcta 35 |||||||||||||||||||||||||||||||||| XL23808 1171 tgggtcatactgtagaacagcttcagttgggatcacaggcttcta 1215 XLRHODOP 36 gggatcctttgggcaaaaaagaaacacagaaggcattctttctat 80 ||||||||||||||||||||||||||||||||||||||||||||| XL23808 1216 gggatcctttgggcaaaaaagaaacacagaaggcattctttctat 1260 XLRHODOP 81 acaagaaaggactttatagagctgctaccatgaacggaacagaag 125 ||||||||||||||||||||||||||||||||||||||||||||| XL23808 1261 acaagaaaggactttatagagctgctaccatgaacggaacagaag 1305 XLRHODOP 126 gtccaaatttttatgtccccatgtccaacaaaactggggtggtac 170 ||||||||||||||||||||||||||||||||||||||||||||| XL23808 1306 gtccaaatttttatgtccccatgtccaacaaaactggggtggtac 1350
unix % water
Smith-Waterman local alignment.
Input sequence: embl:xlrhodop
Second sequence: embl:xl23808
Gap opening penalty [10.0]:
Gap extension penalty [0.5]:
Output file [xlrhodop.water]:
unix % more xlrhodop.water
Local: XLRHODOP vs XL23808 Score: 7448.00 XLRHODOP 2 gtagaacagcttcagttgggatcacaggcttctagggatcctttg 46 ||||||||||||||||||||||||||||||||||||||||||||| XL23808 1182 gtagaacagcttcagttgggatcacaggcttctagggatcctttg 1226 XLRHODOP 47 ggcaaaaaagaaacacagaaggcattctttctatacaagaaagga 91 ||||||||||||||||||||||||||||||||||||||||||||| XL23808 1227 ggcaaaaaagaaacacagaaggcattctttctatacaagaaagga 1271 XLRHODOP 92 ctttatagagctgctaccatgaacggaacagaaggtccaaatttt 136 ||||||||||||||||||||||||||||||||||||||||||||| XL23808 1272 ctttatagagctgctaccatgaacggaacagaaggtccaaatttt 1316 XLRHODOP 137 tatgtccccatgtccaacaaaactggggtggtacgaagcccattc 181 ||||||||||||||||||||||||||||||||||||||||||||| XL23808 1317 tatgtccccatgtccaacaaaactggggtggtacgaagcccattc 1361 XLRHODOP 182 gattaccctcagtattacttagcagagccatggcaatattcagca 226 ||||||||||||||||||||||||||||||||||||||||||||| XL23808 1362 gattaccctcagtattacttagcagagccatggcaatattcagca 1406 XLRHODOP 227 ctggctgcttacatgttcctgctcatcctgcttgggttaccaatc 271 ||||||||||||||||||||||||||||||||||||||||||||| XL23808 1407 ctggctgcttacatgttcctgctcatcctgcttgggttaccaatc 1451 XLRHODOP 272 aacttcatgaccttgtttgttaccatccagcacaagaaactcaga 316 ||||||||||||||||||||||||||||||||||||||||||||| XL23808 1452 aacttcatgaccttgtttgttaccatccagcacaagaaactcaga 1496 XLRHODOP 317 acacccctaaactacatcctgctgaacctggtatttgccaatcac 361 ||||||||||||||||||||||||||||||||||||||||||||| XL23808 1497 acacccctaaactacatcctgctgaacctggtatttgccaatcac 1541
In these cases we have not had to adjust the gap parameters from the defaults used in these programs. You should be aware that you may need to do so with your own sequences.
EMBOSS contains other pairwise alignment programs - stretcher
and matcher are global and local alignment programs respectively that are
less rigorous than needle and water and therefore run
more quickly; they may be useful for database
searching. supermatcher is designed for local alignments of
very large sequences and is even less rigorous in its
implementation. The documentation pages for all these programs can be
found at
http://emboss.sourceforge.net/apps/